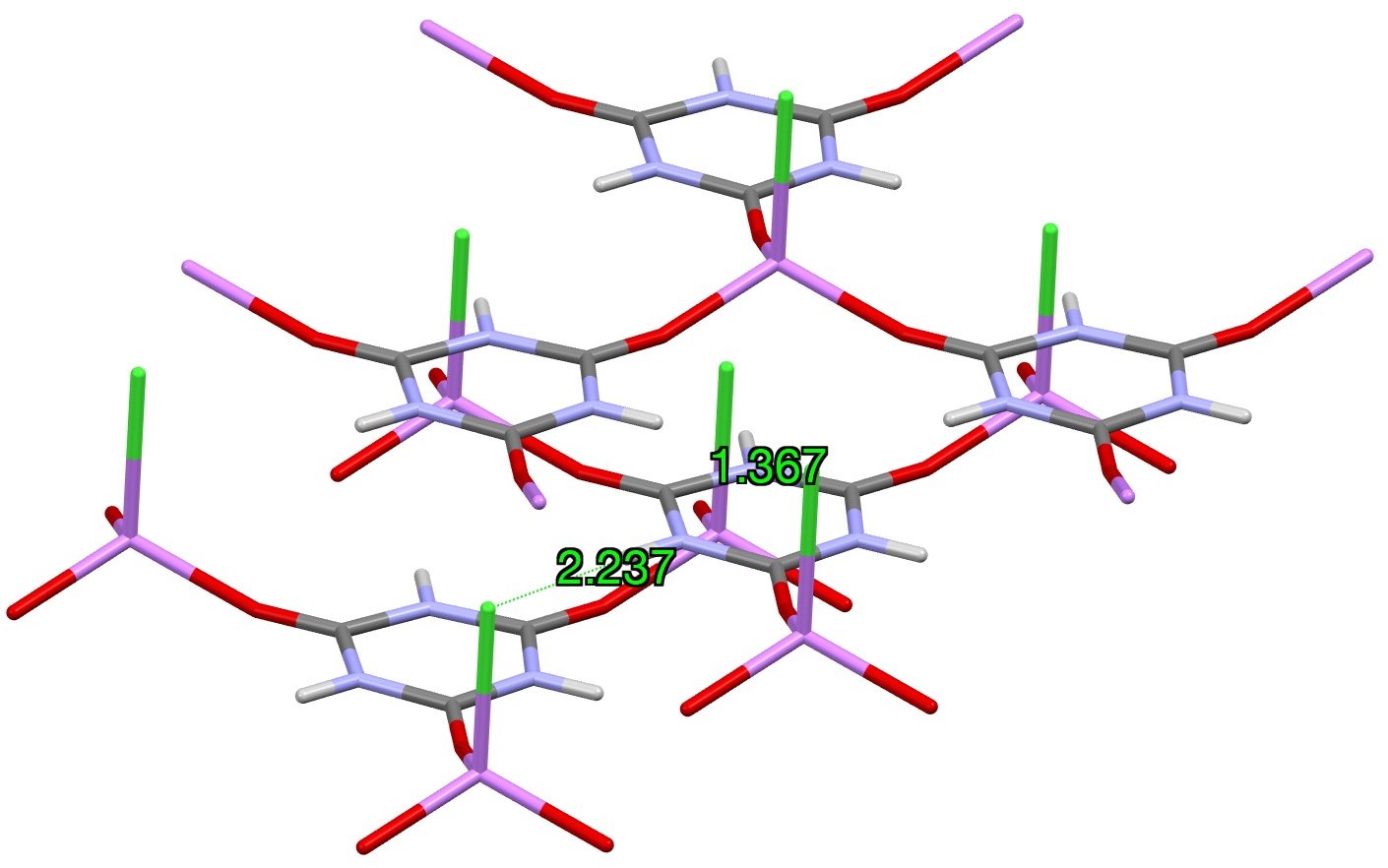
In my story about one of the molecules of the year, cyclo[48]carbon,[cite]10.1126/science.ady6054[/cite] I noted that the DFT method used in the literature to model the C-C bond length alternation around the ring (OX B3LYP30[cite]10.1021/acsnano.4c14100[/cite]) had been re-calibrated against a remeasured crystal structure[cite]10.5517/ccdc.csd.cc2gzmz2[/cite] of C18H18 or [18]-annulene (below) in order to reproduce the observed values for this molecule.

[18]-annulene
A noteworthy aspect of this structure is the six hydrogen atoms pointing into the centre of the ring, which come into very close contact with each other. The conventional method of refining the crystal structure (which includes an assumption that the electron density surrounding the H and indeed other atoms is spherical) results in C-H distances which are too short by about 0.1Å, which has the knock on effect that the H…H separations are now too long. The recent introduction of a refinement method (NoSpherA2) which uses DFT-calculated non-spherical atom electron density distributions rather than spherical ones has the effect of producing more sensible values for e.g. C-H distances[cite]10.59350/5dy8w-0zs92[/cite] and so by implication, results in much shorter inner H..H distances for [18]-annulene. The question now is: do these shorter H…H distances in turn have any effect on the C-C ring distances, and hence affect the alternation of these distances around the ring and the resulting outcome of the calibration process for the development of the OX B3LYP30 method.
Method: We decided to re-refine the structure of [18]annulene (CCDC refcode ANULEN03[cite]10.5517/ccdc.csd.cc2gzmz2[/cite]) using modern quantum crystallography (NoSpherA2[cite]10.1107/S0021889808042726[/cite],[cite],[cite]10.1039/d0sc05526c[/cite]). To do this, we used Def2-SVP as the basis set and wB97X-V for the method, with a multiplicity of 2 in the settings for the OLEX2 program.
The published structure has the molecule sitting across a centre of symmetry so only half of it is unique, and it was found to be disordered with a second orientation of the complete molecule (effectively the macrocycle rotated in plane by ca. 30°) in a ca. 84:16 ratio. This caused trouble with the quantum crystallography refinements as allowing all of the hydrogen atoms to be positionally free (i.e. removing the AFIX commands) and anisotropic at the same time caused 6 of the 8 hydrogen atoms of the minor occupancy component to “wander off” into chemically nonsensical positions, and 4 of the major occupancy plus all 8 of the minor occupancy hydrogen atoms went non positive definite (one of the thermal ellipsoid radii refined to a negative length).
However, we discovered that doing the refinement in stages allowed a more settled structure. Starting with the published structure and allowing all of the hydrogen atoms to be positionally free gave a nice stable result. Allowing the hydrogens to go anisotropic afterwards did result in 1 of the major occupancy and all 8 of the minor occupancy hydrogen atoms going non positive definite (n.p.d.), but the positions of the hydrogen atoms remained sensible. Subsequently reverting all 8 hydrogen atoms of the minor occupancy component to be isotropic resulted in a stable and sensible refinement where the sole non positive definite atom of the major occupancy component corrected itself into being normal (i.e. no longer non positive definite). This is the re-refined version of the structure we used for further analysis below.[cite]10.14469/hpc/15681[/cite]
Analysis: The closest H···H separations for the “inner” hydrogen atoms of the major occupancy orientation emerge as 1.8276(9), 1.8791(9) and 1.9022(8) Ň (Figure 1, mean 1.870Å) This compares to the values extracted from the published structure of 1.99252(4), 2.02490(3) and 2.05217(3), mean = 2.0232Å for the major occupancy orientation, a difference of Δ -0.153Å.

Figure 1.
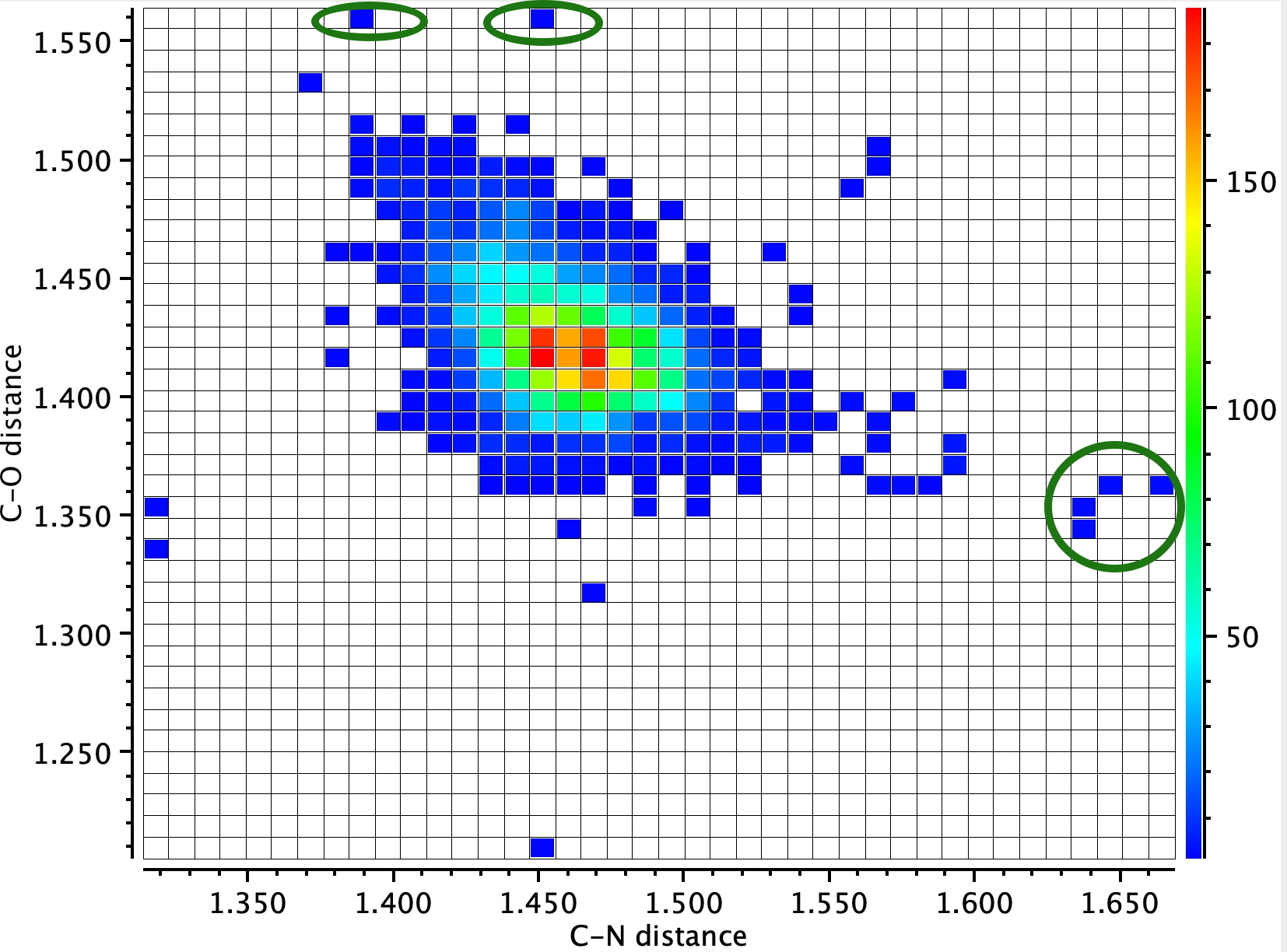
A search for other close H…H contacts: A search of the crystal structure database for close intramolecular H…H distances of <1.9 Å (< 100K, R < 0.05, no errors, excluding H-C-H substructures) reveals the following distribution (Figure 2). Although examples of distances <1.9 Å are relatively sparse (95, February 2026), they are not that unusual. It is highly probable that all these examples were determined using the classical method of spherical atoms. It is to be expected that in the future, examples refined using non-spherical atoms will start appearing – and that one will be specifically able to search for such analyses.

Figure 2.
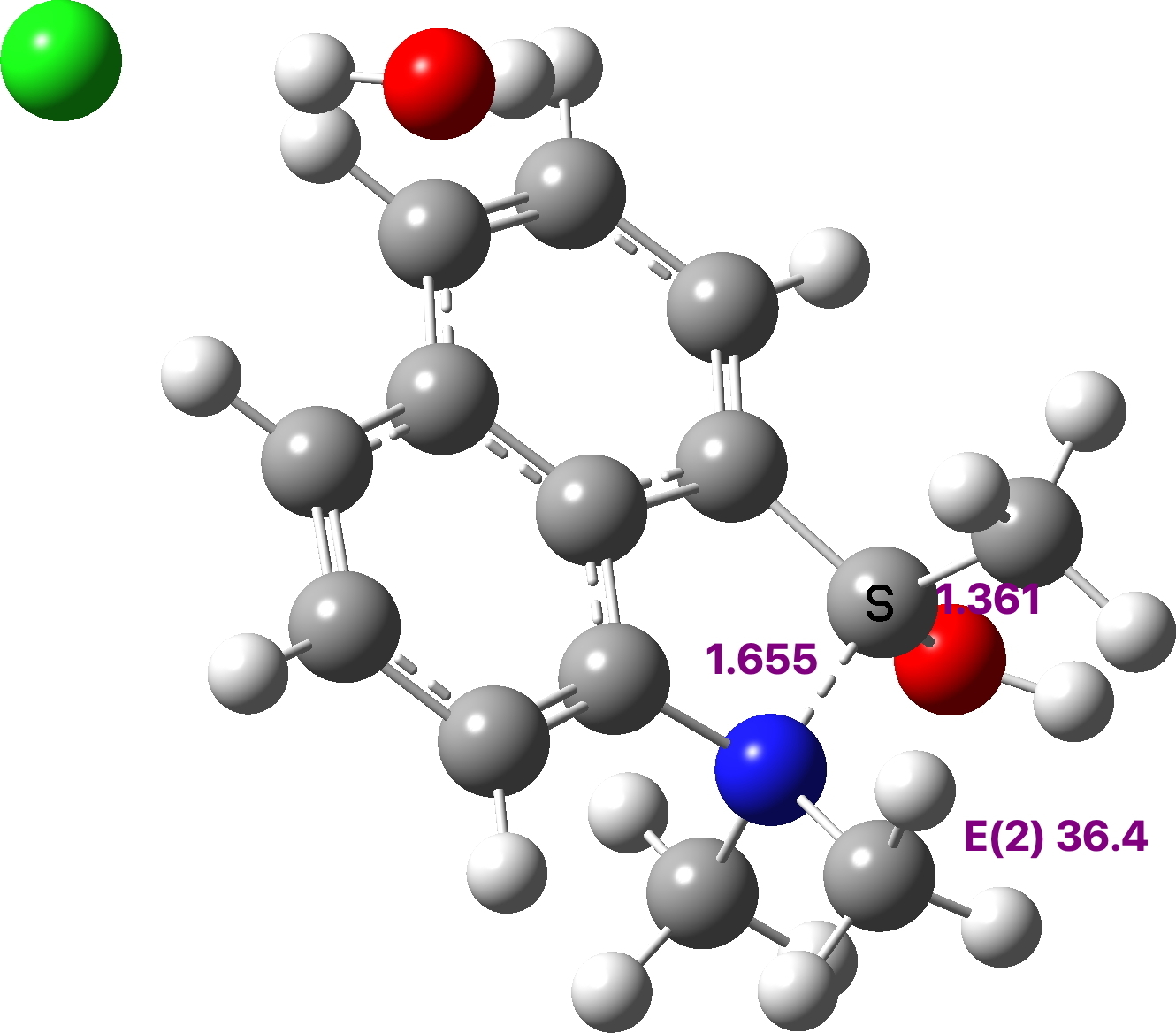
We focussed on just one of the entries in the figure: DOCKEO[cite]10.1021/acs.orglett.9b00717[/cite]
,[cite]10.5517/ccdc.csd.cc21qkbp[/cite] (which is a masked [14]annulene) being an example (see Figure 3) of a compound having an even shorter apparent H…H contact of ~1.65Å (after C-H distance correction). This was also subjected to NoSpherA2[cite]10.1107/S0021889808042726[/cite],[cite],[cite]10.1039/d0sc05526c[/cite] analysis. The structure is in a chiral space group P212121 with no firm indication of the correct enantiomer (the Flack of 0.3(4) is very indeterminate with a large error that encompasses the whole range). It was initially refined as a 2-component racemic twin (using TWIN/BASF) to no real effect. Although this is the standard approach when the Flack is far from zero, it was not really surprising that it had no effect, given the large error (σ = 0.4). Next it was noticed that the original authors had not modelled some evident disorder in one of the CF3 groups. Since the fluorine thermal parameters were reasonable, it is understandable to ignore it, but the largest residual electron density peaks were around this group in obvious disorder positions and with the extra precision desired in quantum crystallography refinements, it was best to model this. A quick rough and ready approach was adopted, one not to be used in a structure of “publication quality”, but enough to “soak up” the electron density. Next, NoSpherA2 was used in a refinement that relaxed the H atom positions (no AFIXes). This worked sensibly, though it had fairly little effect on the R-factor. However, refining the hydrogen atoms anisotropically went poorly; of the 15 hydrogen atoms, 5 went n.p.d, another 5 went nearly n.p.d, and only 2 of them could be described as approaching reasonable. Ultimately, handling the hydrogen atoms was done isotropically. Finally, adding an extinction parameter caused a final 0.2% drop in the R-factor and the H…H distance of closest approach emerged as 1.600Å (Figure 3).

Figure 3.
It is worth noting that this distance is not what it might seem. Thus the calculated DFT H-H distance (using r2SCAN-3c) is 1.8975Å, or Δ0.2975Å. It corresponds to a calculated double minimum potential energy well. However, a Cs-symmetric form with the hydrogen located at the centre of this double well turns out to be a transition state with a shorter H…H separation of 1.7393Å. The imaginary calculated transition mode of νi 61 cm-1 is associated with a tiny free energy barrier of ~0.03 kcal/mol, well below a quantum of vibrational energy and hence the observed hydrogen will in fact correspond to that of a single minimum potential well. The lesson learnt from this analysis is that measured distances (for a single potential well) and calculated distances (for a double potential well) may not always correspond and care must be taken in interpreting such distances.
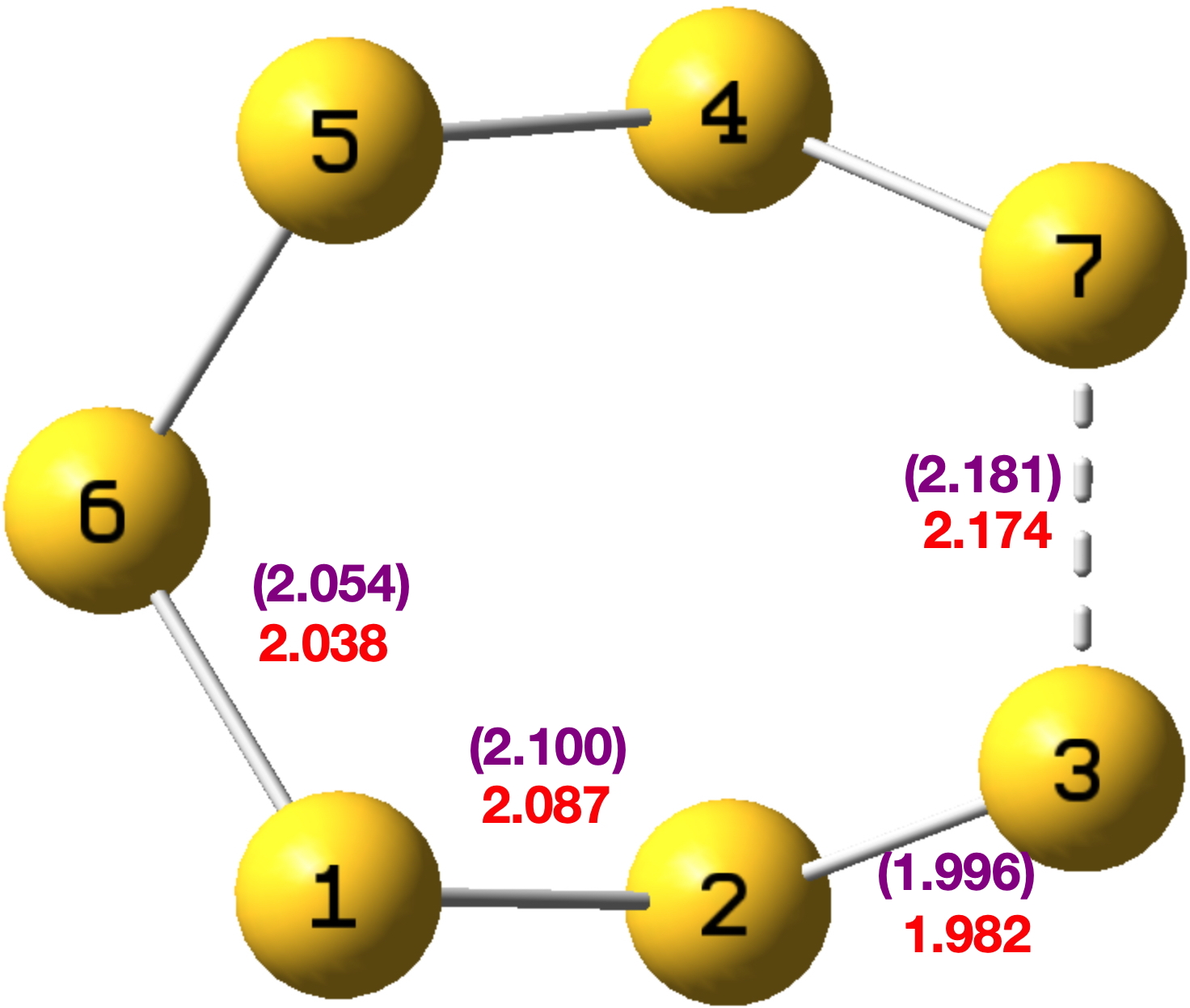
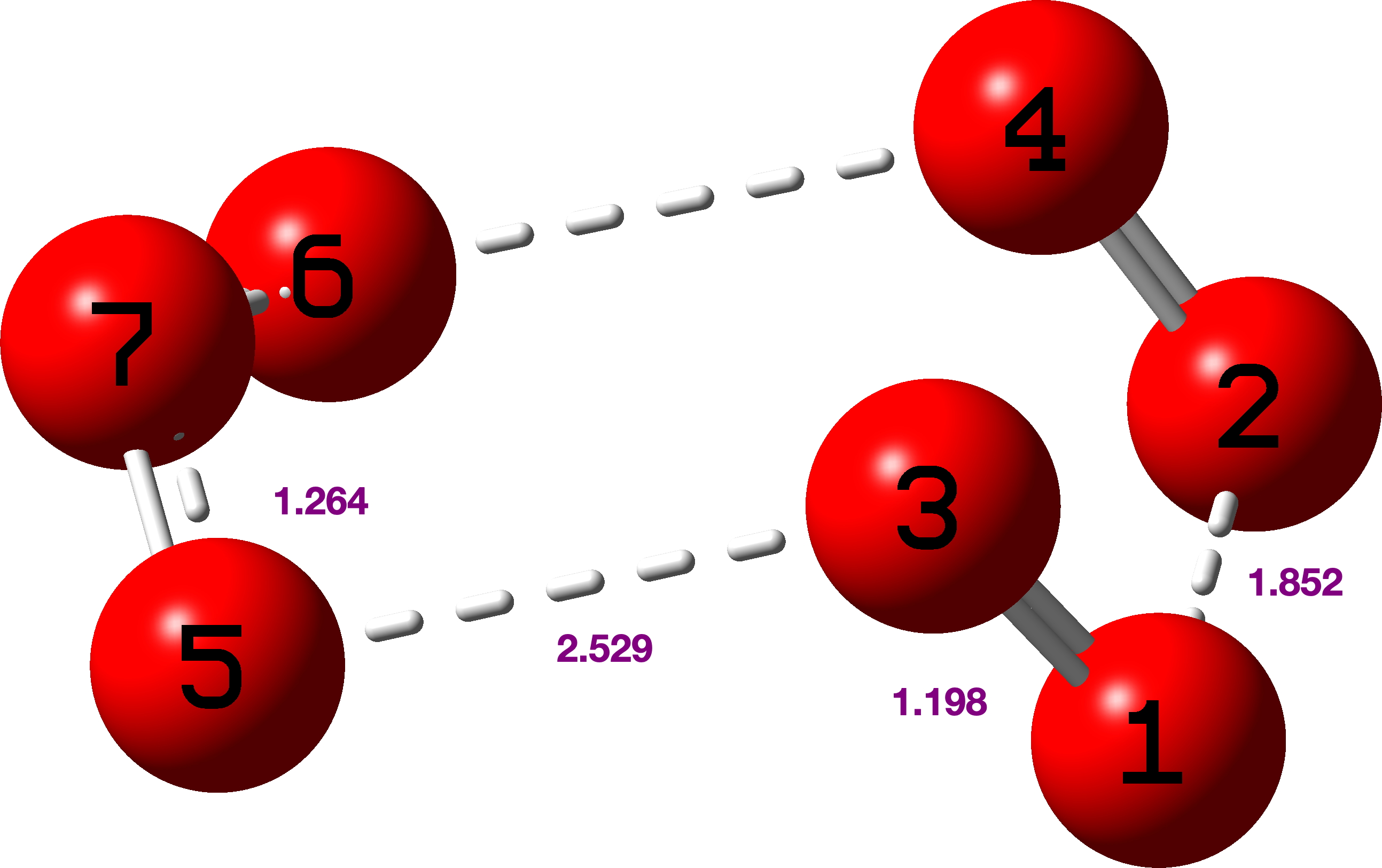
C-C Distances in [18]-annulene. The nine‡ unique pairs of C-C distances in the measured structure of [18]annulene derive from Figure 4 and the atom numbering shown there.

Figure 4.
Table 1. Crystallographic C-C bond lengths and Δ differences for [18]-annulene, Å
Original refinement[cite]10.5517/ccdc.csd.cc2gzmz2[/cite]
old Δ
C2 C3 1.403(2) C2 C1 1.3883(17) 0.0147
C3 C4 1.3913(14) C2 C3 1.403(2) 0.0117
C5 C4 1.3926(15) C3 C4 1.3913(14) 0.0013
C5 C6 1.4056(18) C5 C4 1.3926(15) 0.0130
C7 C6 1.3870(15) C6 C5 1.4056(18) 0.0186
C8 C7 1.3927(15) C7 C6 1.3870(15) 0.0057
C8 C9 1.4032(19) C8 C7 1.3927(15) 0.0105
C9 C1 1.3897(15) C8 C9 1.4032(19) 0.0135
C2 C1 1.3883(17) C9 C1 1.3897(15) 0.0014
Mean 0.0100Å
NoSpherA2 refinement New Δ old Δ
C2 C3 1.4036(12) C2 C1 1.3948(11) 0.0088 0.0147
C3 C4 1.3954(9) C2 C3 1.4036(12) 0.0082 0.0117
C5 C4 1.3939(10) C3 C4 1.3954(9) 0.0015 0.0013
C5 C6 1.4064(11) C5 C4 1.3939(10) 0.0125 0.0130
C7 C6 1.3928(10) C5 C6 1.4064(11) 0.0136 0.0186
C8 C7 1.3938(10) C7 C6 1.3928(10) 0.0010 0.0057
C8 C9 1.4126(12) C8 C7 1.3938(10) 0.0188 0.0105
C9 C1 1.3846(10) C8 C9 1.4126(12) 0.0280 0.0135
C2 C1 1.3948(11) C9 C1 1.3846(10) 0.0102 0.0014
Mean 0.0114Å 0.0100Å
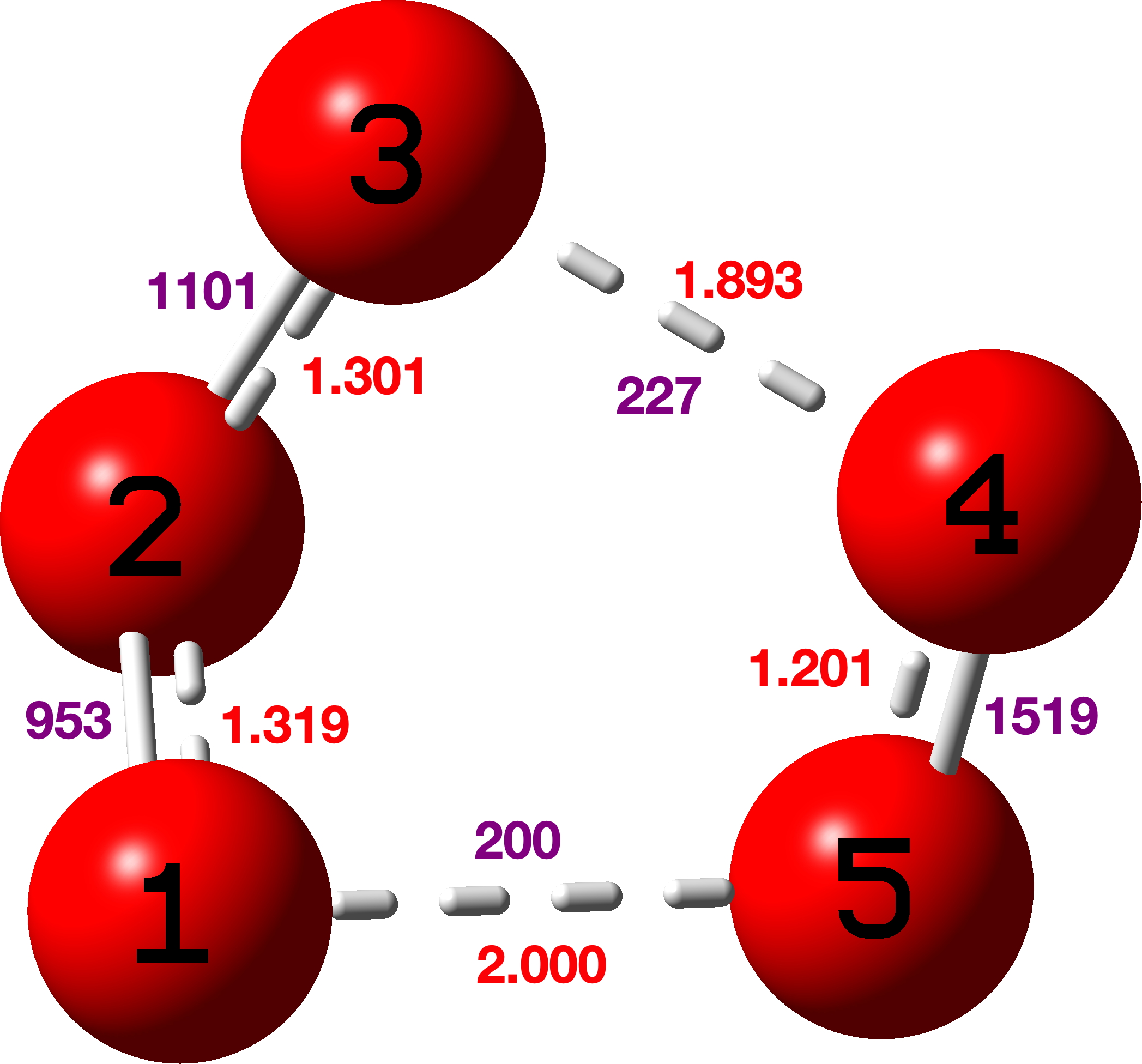
Shown below is the atom numbering used in the r2-SCAN-3C DFT geometry optimisation[cite]10.14469/hpc/15615[/cite] (Figure 5).

Figure 5
Table 2. Computed r2-SCAN-3c C-C bond lengths and Δ differences for [18]-annulene, Å
Δ
1 5 1.40751 5 11 1.39010 0.01741
5 11 1.39010 11 3 1.39020 0.00010
11 3 1.39020 3 15 1.40755 0.01735
3 15 1.40755 15 13 1.39017 0.01738
15 13 1.39017 13 7 1.38998 0.00019
13 7 1.38998 7 9 1.40748 0.01750
7 9 1.40748 9 35 1.39009 0.01739
9 35 1.39009 35 19 1.39009 0.00000
35 19 1.39009 19 23 1.40752 0.01743
Mean 0.0116
Conclusions: We set out to study the extent to which the C-C distances in the [18]-annulene molecule, as used to calibrate a modified DFT method[cite]10.1126/science.ady6054[/cite] could be affected by steric compressions in the centre of the ring caused by close approaches of the inward pointing hydrogens. The NoSpherA2 method of crystal structure refinement results in a slight increase in the C-C bond length alternation around the ring, from 0.0100 to 0.0114Å, but given that this analysis is quick and easy to perform, there is no reason not to use it as a standard method for structures used for calibration purposes. The newly re-refined bond alternating distance compares with 0.0116Å calculated using the r2-SCAN-3c DFT procedure and 0.0112Å calculated using the original literature[cite]10.1126/science.ady6054[/cite] OX B3LYP30 method which had been calibrated against this distance. Both the DFT methods are thus seen to perform very well against the measured bond length alternation. Clearly however there is a need to undertake more such studies for a clearer understanding of the performance of DFT methods in this area.
‡As the macrocycle sits across a centre of symmetry there are only 3 unique H…H distances and 9 unique C-C differences.

 Here I take a look at this very unusual structure, the core of which is an imidazole ring coordinated via nitrogen to the metal Cu.
Here I take a look at this very unusual structure, the core of which is an imidazole ring coordinated via nitrogen to the metal Cu.
 reveals 12 hits, with a range of O-O distances ranging from 1.37 to 1.54Å. A histogram of the O-O lengths in the Cu(O-O)Cu sub structure shown below shows quite a distribution amongst the 12 known examples.
reveals 12 hits, with a range of O-O distances ranging from 1.37 to 1.54Å. A histogram of the O-O lengths in the Cu(O-O)Cu sub structure shown below shows quite a distribution amongst the 12 known examples.





























{kind=link}
{kind=link}