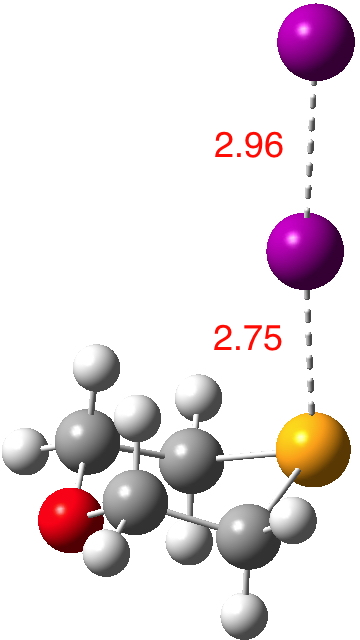
Following the general recognition of carbon as being tetrahedrally tetravalent in 1869 (Paterno) and 1874 (Van’t Hoff and Le Bell), an early seminal exploitation of this to the conformation of cyclohexane was by Hermann Sachse in 1890.[cite]10.1002/cber.189002301216 [/cite] This was verified when the Braggs in 1913[cite]10.1098/rspa.1913.0084[/cite], followed by an oft-cited article by Mohr in 1918,[cite]10.1002/prac.19180980123[/cite] established the crystal structure of diamond as comprising repeating rings in the chair conformation.† So by 1926, you might imagine that the shape (or conformation as we would now call it) of cyclohexane would be well-known. No quite so for everyone!
When The Journal of the Imperial College Chemical Society (Volume 6) was brought to my attention, I found an article by R. F Hunter;
He proceeds to argue as follows:
- The natural angle subtended at a tetrahedral carbon is 109.47°.
- “The internal angle between the carbon to carbon valencies of a six-membered ring cyclohexane will, if the ring is uniplanar, be … 120°.
- “When the cyclohexane ring is prepared … we must therefore have the pushing apart of two of the valencies”.
- The object of the experiments commenced in this College in 1914 was “to find what effect the pushing apart of the valencies …must have on the angle between the remaining pair of valencies“.
- You do wonder then why the assumption highlighted in red above was never really questioned during the twelve-year period of investigating angles around tetrahedral carbon.
The article itself is quite long, reporting the synthesis of many compounds in search of the postulated effect. Of course around twenty years later, Derek Barton used the by then generally accepted conformation of cyclohexane to explain reactivity in what become known as the theory of conformational analysis.
These two articles dating from 1926, and probably thought lost to science, show how some ideas can take decades to have any influence, whilst others can take root very much more quickly.
Postscript added in 2022. In the news recently has been Lonsdaleite, another allotrope of carbon. The saturated diamond structure has all the tetrahedral carbon forming six-membered rings in the chair† conformation. In Lonsdaleite,[cite]10.1063/1.1841236[/cite] there are boat as well as chair rings!


†The chair cyclohexane structure is easily discerned from Figure 7 in the Braggs’ paper![cite]10.1098/rspa.1913.0084[/cite]