Members of the chemical FAIR data community have just met in Orlando (with help from the NSF, the American National Science Foundation) to discuss how such data is progressing in chemistry. There are a lot of themes converging at the moment. Thus this article[cite]10.1039/c7np00064b[/cite] extolls the virtues of having raw NMR data available in natural product research, to which we added that such raw data should also be made FAIR (Findable, Accessible, Interoperable and Reusable) by virtue of adding rich metadata and then properly registering it so that it can be searched. These themes are combined in another article which made a recent appearance.[cite]10.1021/acsomega.8b03005[/cite]
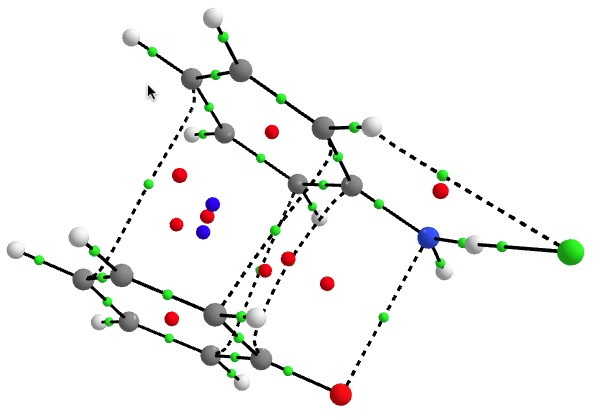
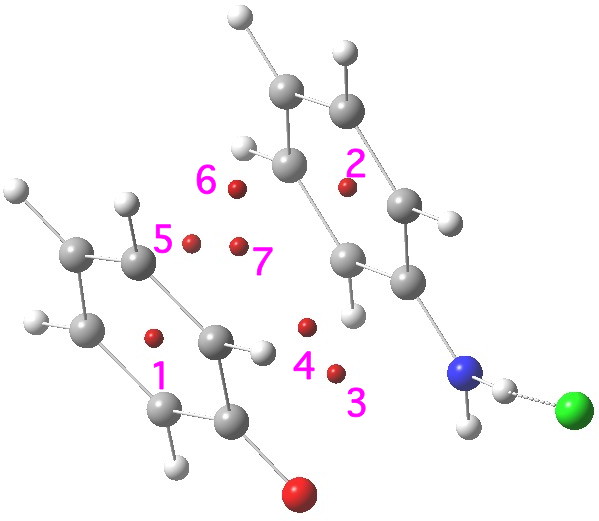
One of the speakers made a very persuasive case based in part on e.g. the following three molecules which are discussed in the first article[cite]10.1039/c7np00064b[/cite] (the compound numbers are taken from there). The question was posed at our meeting: why did the referees not query these structures? And the answer in part is to provide referees with access to the full/primary/raw NMR data (which almost invariably they currently do not have) to help them check on the peaks, the purity and indeed the assignments. I am sure tools that do this automatically from such supplied data by machines on a routine basis do exist in industry (and which is something FAIR is designed to enable). Perhaps there are open source versions available?
| 17 | 18 | 19 |
|---|---|---|
 |
 |
 |
| 328[cite]10.1002/ejoc.201301308[/cite] | 348 | 713 |
Here I suggest a particularly simple and rapid “reality check” which I occasionally use myself. This is to compute the steric energy of the molecule using molecular mechanics. The mechanics method is basically a summation of simple terms such as the bond length, bond angle, torsion angle, a term which models non bonded repulsions, dispersion attractions and electrostatic contributions. The first three are close to zero for an unstrained molecule (by definition). The last three terms can be negative or positive, but unless the molecule is protein sized, they also do not depart far from zero. A suitable free tool that packages all this up is Avogadro.
The procedure is as follows
- Start from the Chemdraw representation of the molecule. If the publishing authors have been FAIR, you might be able to acquire that from their deposited data. Otherwise, redraw it yourself and save as e.g. a molfile or Chemdraw .cdxml file.
- Drop into Avogadro, which will build a 3D model for you using stereochemical information present in the Chemdraw or Molfile.
- In the E tool (at the top on the left of the Avogadro menu) select e.g. the MMFF94 force field. This is a good one to use for “organic” molecules for which the total steric energy for “normal” molecules is likely to be < 200 kJ. Calculate that for your system; this normally takes less than one minute to complete. The values obtained for the three above are shown in the table. All three are well over 200 kJ/mol, which should set alarm bells ringing.
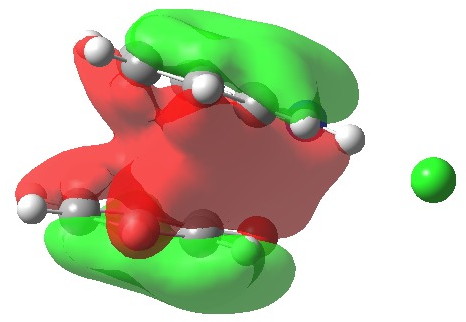
- A “more reasonable” structure for 17 is shown below. This has a steric energy of 152 kJ/mol, some 176 kJ/mol lower than the original structure. This does not of itself “prove” this alternative, but it is a starting point for showing it might be correct.
 Of course mis-assigned but otherwise reasonable structures are unlikely to be revealed by the steric energy test. But impossible ones will probably always be flagged as such using this procedure.
Of course mis-assigned but otherwise reasonable structures are unlikely to be revealed by the steric energy test. But impossible ones will probably always be flagged as such using this procedure.
Postscript: Hot on the heels of writing this, the molecule Populusone came to my attention.[cite]10.1021/acs.orglett.9b00423[/cite] On first sight, it seems to have some of the attributes of an “impossible molecule” (click on diagram below for 3D coordinates).

However, it has been fully characterised by x-ray analysis! The steric energy using the method above comes out at 384 kJ/mol, which in the region of impossibility! This can be decomposed into the following components: bond stretch 30, bend 51, torsion 32, van der Waals (including repulsions) 177, electrostatics 87 (+ some minor cross terms). These are fairly evenly distributed, with internal steric repulsions clearly the largest contributor. The C=C double bond is hardly distorted however, which is in its favour. Clearly a natural product can indeed load up the unfavourable interactions, and this one must be close to the record of the most intrinsically unstable natural product known!
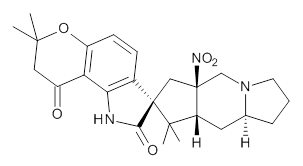 But there was one aspect of this that I did want to have a confidence level for; the absolute configuration of citrinalin B. Reading the article Steve quotes[cite]10.1038/nature13273[/cite], one sees this aspect is attributed to ref 5[cite]10.1021/jo051499o[/cite], dating from 2005. There the configuration was assigned on the basis of “comparison of the electronic circular dichroism (ECD) spectra for 1 and 2 with those of known spirooxiindole alkaloids“. However, this method can fail[cite]10.1002/chem.201101129[/cite]. Also, one finds “comparison of the vibrational circular dichroism (VCD) spectra of 1 with those of model compounds“[cite]10.1021/jo051499o[/cite]. Nowadays, one would say that there is no need for model compounds, why not measure and compute the VCD of the actual compound? Even a determination using the Flack crystallographic method can occasionally be wrong![cite]10.1021/jo401316a[/cite]. Which leads to asking what typical confidence levels might be for these three techniques, and indeed whether improving instrumentation means that the confidence level gets higher with time. OK, I am going to guess these.
But there was one aspect of this that I did want to have a confidence level for; the absolute configuration of citrinalin B. Reading the article Steve quotes[cite]10.1038/nature13273[/cite], one sees this aspect is attributed to ref 5[cite]10.1021/jo051499o[/cite], dating from 2005. There the configuration was assigned on the basis of “comparison of the electronic circular dichroism (ECD) spectra for 1 and 2 with those of known spirooxiindole alkaloids“. However, this method can fail[cite]10.1002/chem.201101129[/cite]. Also, one finds “comparison of the vibrational circular dichroism (VCD) spectra of 1 with those of model compounds“[cite]10.1021/jo051499o[/cite]. Nowadays, one would say that there is no need for model compounds, why not measure and compute the VCD of the actual compound? Even a determination using the Flack crystallographic method can occasionally be wrong![cite]10.1021/jo401316a[/cite]. Which leads to asking what typical confidence levels might be for these three techniques, and indeed whether improving instrumentation means that the confidence level gets higher with time. OK, I am going to guess these.