The title of this post summarises the contents of a new molecular database: www.molecularspace.org[cite]10.1021/jz200866s[/cite] and I picked up on it by following the post by Jan Jensen at www.compchemhighlights.org (a wonderful overlay journal that tracks recent interesting articles). The molecularspace project more formally is called “The Harvard Clean Energy Project: Large-scale computational screening and design of organic photovoltaics on the world community grid“. It reminds of a 2005 project by Peter Murray-Rust et al at the same sort of concept[cite]10.1007/s00894-005-0278-1[/cite] (the World-Wide-Molecular-Matrix, or WWMM[cite]10.1186/1758-2946-3-42[/cite]), although the new scale is certainly impressive. Here I report my initial experiences looking through molecularspace.org
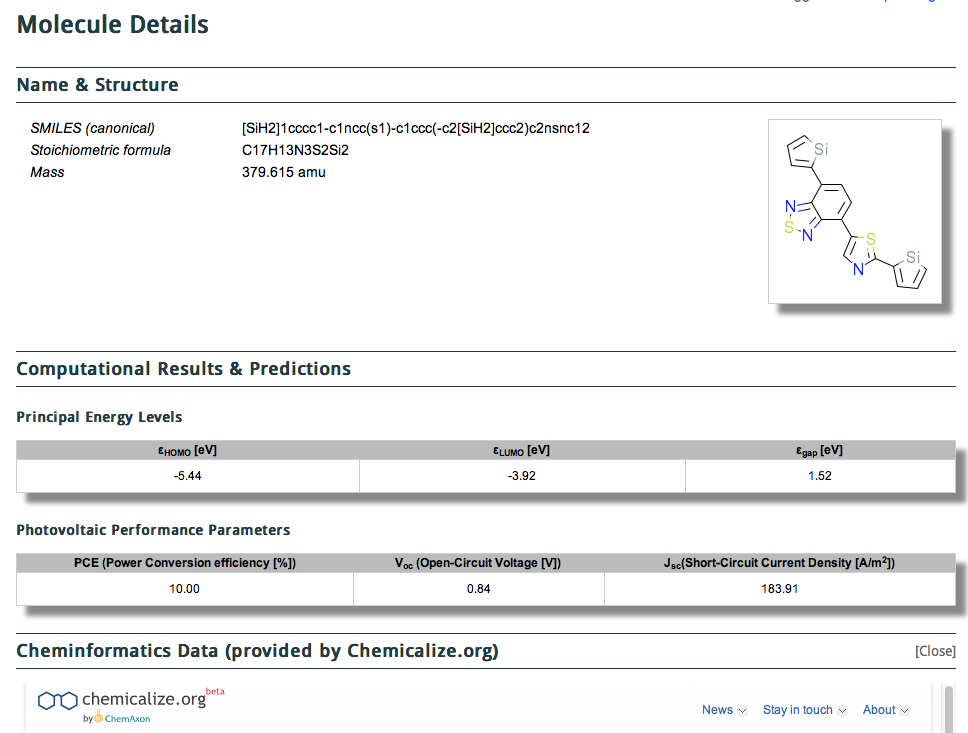
The 150,000,000 calculations are released under the the CC-BY license, which is an encouraging (open) start. One does need however to login to the site, which I was able to do using my Google credentials. Shown below is a screenshot of a typical result in a search (of Power conversion efficiency in my case).
It comes in two parts, the first being the structure (given as a SMILES and 2D layout) with the principle predicted energy levels and predicted photovoltaic performance listed below that. This is then followed by what might be called an annotation with further computed/predicted properties using the algorithms applied by Chemicalize.org. This idea that a data set could accrete via semantically powerful annotations using other tools was also very much part of the concept of the WWMM (the matrix had at its heart a molecule in one dimension and a property, measured or computed in the other. The matrix is of course very sparse, which is why it needs annotation!).
It was at this point however that I started to wonder how I might add other annotations, based perhaps on other types of calculations. But thus far at least, I have not found any trace of something which I could immediately use for my own calculation; 3D coordinates specifically. Thus, the HOMO-LUMO energy gap is the key property which makes molecularspace unique and valuable (to someone working in the field of photovoltaics). But HOMO/LUMO gaps can be calculated in many different ways, and it can always be valuable to calibrate/validate the reported values against other methods. Perhaps if I continue to look, I might find these 3D coordinates (which, for 2,300,000 molecules would be a very valuable resource). Certainly for example, should I wish to do so, I could not at the moment readily replicate the calculation for any specific entry on the molecularspace site (which can be regarded as an essential component of scientific validation). When I use the first person, I mean of course either myself as a human or a software agent acting on my behalf (the latter having the endurance to repeat its procedures millions of times if necessary).
The reader of this blog may have noticed that whenever I report a calculation here, I like to cite its doi (more formally its handle), which links to a digital repository. In my case, the repository certainly carries the 3D coordinates, and also the full wavefunction provided if the reader wishes other properties to be derived from it.‡ Now if molecularspace is able to provide that in the fullness of time, it truly would be an impressive resource.
But the important take-home message from molecularspace is that archiving (under a CC-BY license) the “big” data from any given research in a manner which makes it readily re-usable by others (perhaps from quite different fields of science) is now an essential requisite of doing science. And it is really nice to see good examples of this in practice!
‡ Generally, the calculations I perform for this blog are published in a DSpace repository (the original one, started in 2006[cite]10.1021/ci7004737[/cite]), and more recently in Chempound (a project by Peter Murray-Rust and colleagues which emerged out of the WWMM experiments) as well as Figshare[cite]10.6084/m9.figshare.739310[/cite]. The first and the third assign unique handles (i.e. a doi) to the data; chempound does not (and neither does molecularspace).
The iconic diagram below represents a cornerstone of organic chemistry. Generations of chemists have learnt early on in their studies of the subject that these two representations of where the electron pairs in benzene might be located (formally called electronic resonance or valence bond forms) each contribute ~50% to the overall wavefunction, and that the real electronic description is in effect an average of these two (that is the implied meaning of the double headed arrow). This means that the six C-C bonds in benzene must all be of equal length. The diagrams, everyone knows, do not mean that benzene has three short and three long C-C bonds.
The Kekulé structures of benzene. Click for 3D.The diagram has much other implied semantics. Thus there is no explicit three dimensional information; the molecule looks (and is) flat, and it is tempting to conclude that the electrons are flat and two dimensional as well. Indeed, up to around 1930 (some 105 years after its first discovery), the electrons in benzene were always represented as all lying in the plane of the molecule. This changed when Hückel announced the principle of σ/π separation. These were the labels he gave to two different symmetries of electrons (actually derived for ethene), one set which did genuinely occupy the plane of the molecule, and a second (π) set for which this plane represented a node (a region of zero probability for the electron density). The π electrons could instead be regarded as occupying the space above and below that plane. Hückel went on to develop a quantum mechanical theory for benzene based purely on those π-electrons, of which there are six. This (now called Hückel) theory predicted that the averaged structure noted above emerged naturally, along with another concept known as π-electron resonance energy. This is the difference in energy between the symmetric form of benzene and a structure in which the six π electrons do not interact as a whole, but which are localized into three pairs located in the regions of the double bonds. Most people interpret this latter as being equivalent to the two Kekulé forms shown above. Symmetrizing the structure (from D3h to the higher D6h symmetry) is accompanied by reducing the π-energy of the system by that resonance term (often estimated as around -152 kJ/mol of stabilization). For benzene in other words, this is the difference in energy between the symmetric species and a (hypothetical) bond localized cyclohexatriene.
With such a focus on the π-electrons, it seemed natural to accept that the reason why benzene has six equal C-C lengths is because of the resonance energy gained by the π-electrons when adopting the six-fold symmetric form. Prior to around 1961, no-one would have dissented from that point of view. The first to do so was Berry (see DOI: 10.1063/1.1732256 ), but his was a lone voice at that time. But mysterious and inexplicable observations started to come to light. Perhaps the most direct was a study of the excited state of benzene, in which one π-electron is promoted from a bonding to a higher energy and antibonding π-orbital (known as a π-π* excitation, see DOI 10.1063/1.435193). A schematic illustration of this process is shown below.
The Hückel Molecular orbital picture for ground and excited states of benzene
Diagram (a) shows the normal population of electrons in the (three lowest) energy levels derived using Hückel’s theory. Diagram (b) shows how this changes in the first excited singlet state, which would be expected to have weaker π-bonds. The vibrational spectrum of a molecule is one way of measuring how strong the bonds in a molecule are. Berry had already implied that one particular vibrational mode, the so-called Kekulé mode (also known as the b2u mode using group theory) seemed unusually low in frequency. In other words, this distorsion was easier than it should have been, and this Berry attributed to the (then almost heretical view) that the π-electrons did not in fact promote a hexagonal form of benzene. This was instead induced by the σ electrons, which occupy the plane of the molecule. This effect prevailed over the π-electrons, which were in fact trying to get benzene to adopt a bond-alternating geometry (managing instead only to lower the energy of the b2u mode). When the vibrational spectrum of the excited state of benzene was analyzed in 1977, it appeared to spectacularly vindicate Berry (DOI 10.1063/1.435193). The Kekulé mode has a value of 1309 cm-1 for the normal ground state of benzene, but an exalted value of 1570 cm-1 in the excited state. This means that as the bonding due to the π-electrons is weakened by placing one of them in an antibonding orbital, their overall ability to distort the geometry is also weakened. As a result, the resistance to such distorsion (the Kekulé mode) is in turn strengthened by an amount corresponding to +261 cm-1. It was evidence such as this, and much else besides that Shaik and his co-workers used to promote the idea of π-distortivity in benzene (DOI: 10.1021/cr990363l). Despite such advocacy, the idea that all the six bonds in benzene are equal despite rather than because of the π-electrons is still rarely taught in introductory organic chemistry.
But the story of excited benzene is not yet quite finished! In 2006, Blancafort and Sola (DOI: 10.1021/jp064885y) reminded us that the (1B2u) excited state of benzene exhibits a type of geometrical distorsion known as pseudo Jahn-Teller (PJT), the origins of which have nothing to do with any of the previous arguments. The effect instead arises because the promoted electron emerges from a so-called energy degenerate orbital, and jumps into another degenerate orbital (Figure b above). The exaltation of the b2 vibrational mode is in fact strongly coupled with this PJT effect, which complicates disentangling the two effects (PJT and π-distortivity).
So another excited state is here proposed which is not susceptible to the PJT effect. Figure (c) above shows a π-quintet state in which two electrons are both promoted to anti-bonding orbitals. Now the π-electron bonding has been well and truly weakened! When the vibrational modes are calculated for the (D6h-symmetric) geometry of benzene at the same level of theory (B3LYP/aug-cc-pvtz) for both singlet ground state and quintet excited state one finds that the b2u vibrational mode has the value of 1332 cm-1 for the former and 1524 cm-1 for the latter. Significantly, the former mode shows a contribution to the motion from the hydrogen atoms. These, being light, tend to increase the wavenumber of the vibrational mode. The same mode in the quintet state however shows motion of the carbon atoms only (Click on the diagram below to view the b2u mode for the quintet state of benzene, and note how little motion of the hydrogen atoms there is). It is a pure Kekulé mode, whereas that for the ground state is not! If the motion of the hydrogens in the ground state of benzene is suppressed by artificially changing the atomic weight of the hydrogen in the mass-weighting scheme to a large value, the calculated b2u vibrational mode drops to around 1317 cm-1. This means the quintet state mode of benzene is exalted by 207 cm-1, and being PJT-free, it is a truer reflection of the effect of the π-electrons. Thus the effect first speculated upon by Berry, and championed by Shaik is spectacularly vindicated (again!).
The b2u modes in benzene for (a) ground state and (b) quintet state. Click for 3D.But what of the title for this post? Well, the C-C length in the singlet ground state of benzene is 1.391Å. In the quintet state, it becomes longer at 1.454Å (which is almost exactly the value that Berry originally suggested should be used for the hypothetical cyclohexatriene geometry). Despite this lengthening, the Kekulé mode clearly gets stronger. Why is this noteworthy? Well, it is almost always assumed that if a bond is shorter, it means stronger. In this case, we have an example of six bonds each getting shorter and weaker (at least as measured by the b2u mode of vibration), or as the title states, longer and stronger in the quintet state of benzene. Oh, and what about that π-resonance energy which we started with? Does it play no role after in the symmetric structure of benzene? Well, in fact it does! The answer is that the π-resonance energy is still at its maximum stabilization at the hexagonal structure of benzene, but it is the total π-energy that achieves its maximum stability at the non-hexagonal structure. These two energies are quite different beasts, and they each prefer a different geometry!
Acknowledgments
This post has been cross-posted in PDF format at Authorea.