Sodium borohydride is the tamer cousin of lithium aluminium hydride (LAH). It is used in aqueous solution to e.g. reduce aldehydes and ketones, but it leaves acids, amides and esters alone. Here I start an exploration of why it is such a different reducing agent.

Initially, I am using Li, not Na (X=Li), to enable a more or less equal comparison with LAH, with water molecules to solvate rather than ether (n=2,3,5) and R set to Me. First, n=2, for which the IRC is shown below. In this model, we will assume that the carbonyl has not first reacted with water to form a gem-diol. The free energy barrier is 9.6 kcal/mol (ωB97XD/6-311+G(d,p)/SCRF=water) which corresponds to a very fast reaction at room temperatures.
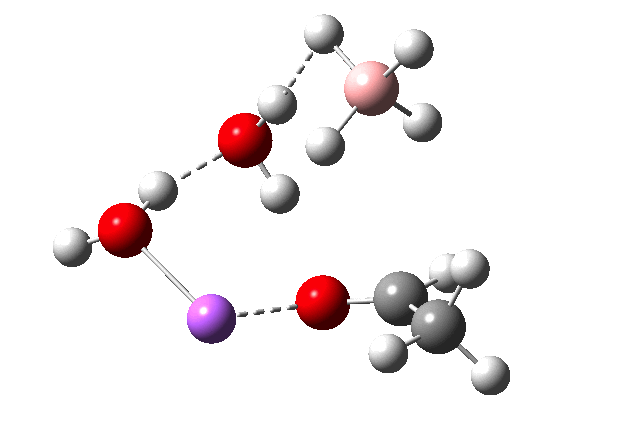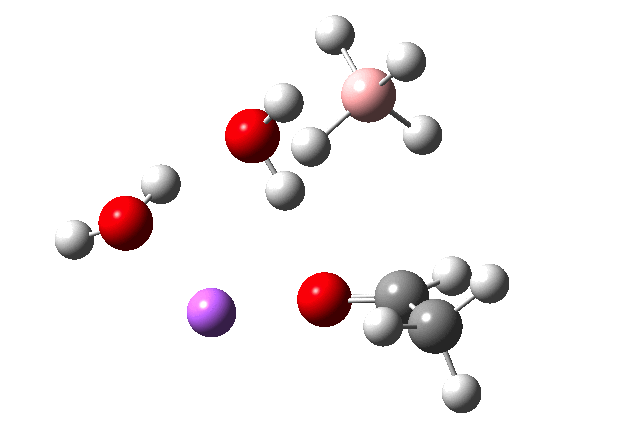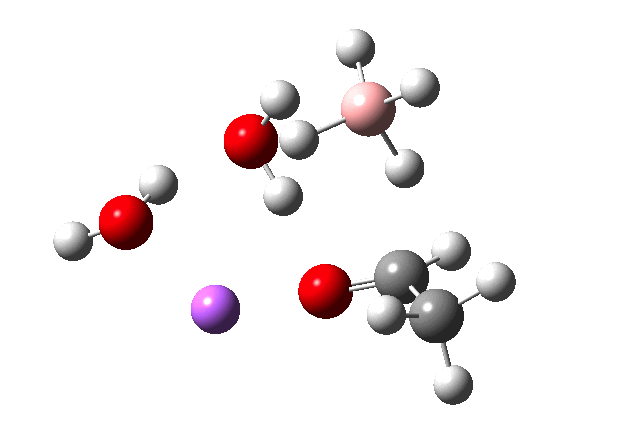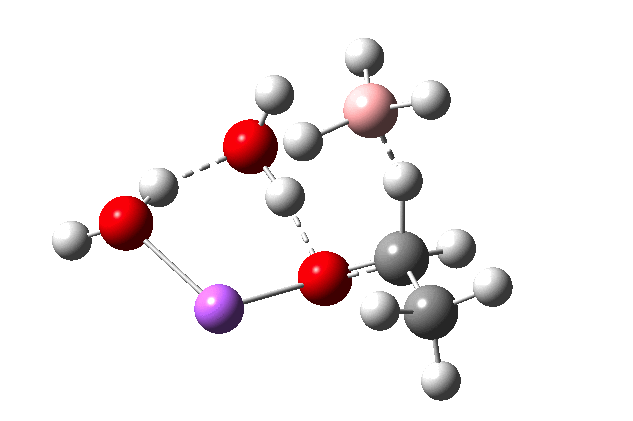
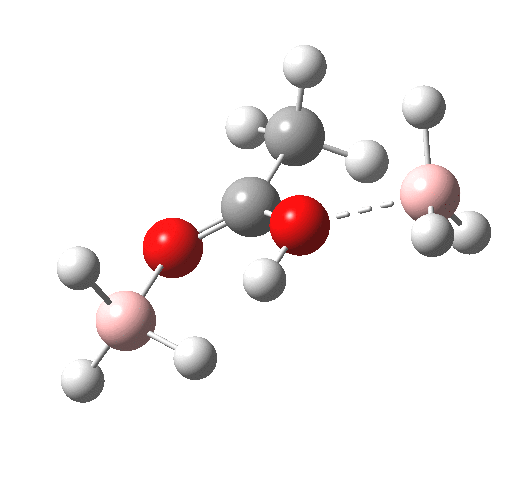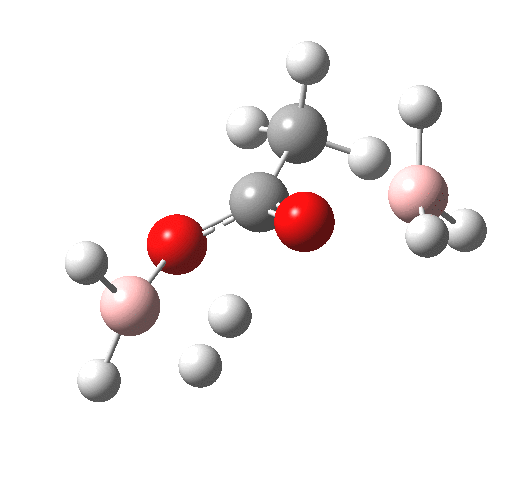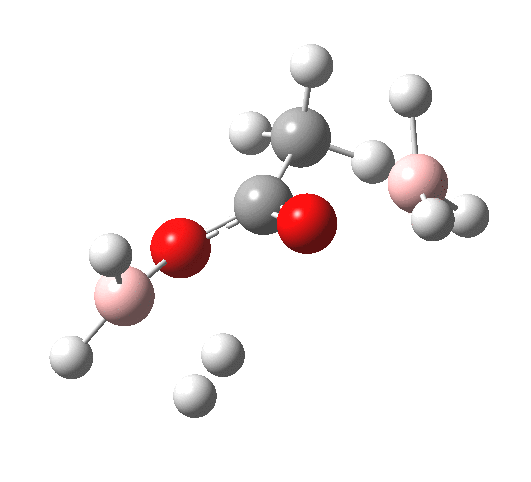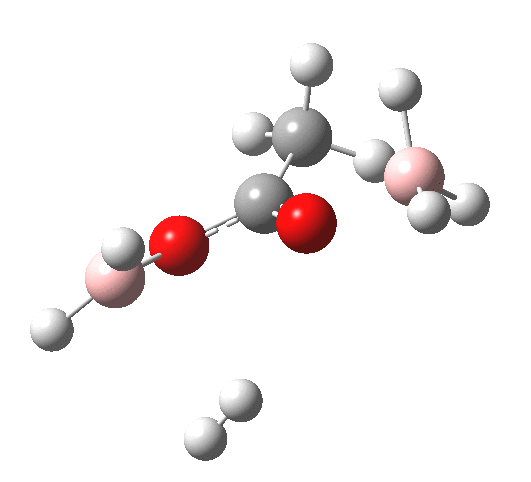
The immediate product is, if anything, more interesting than the transition state[cite]10.14469/ch/191186[/cite] with quite a stretched length for the newly formed C-H bond and predicted stretching wavenumber for this bond of 2137 cm-1. This effect is similar to that seen for the LAH reduction of cinnamaldehyde, and is due to stereoelectronic antiperiplanar alignment of the oxyanionic oxygen lone pair with the C-H bond. This species is also some 6.5 kcal/mol higher in energy than the reactant, and is clearly not the final product of the reaction (which must contain e.g. B-O bonds), the mechanism for which will not be investigated here immediately.
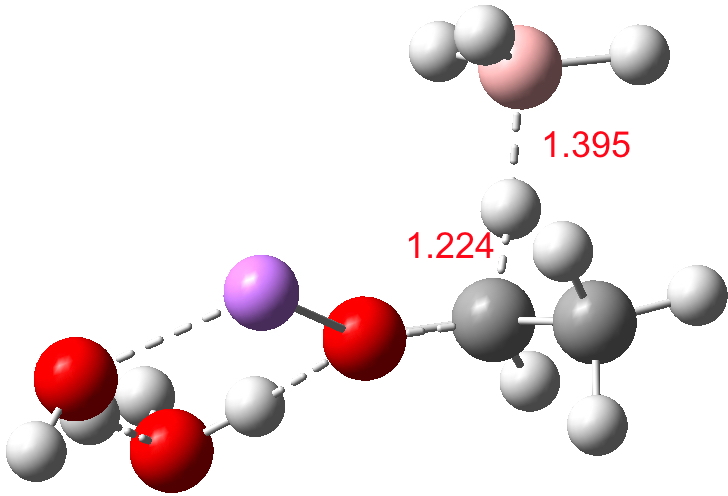
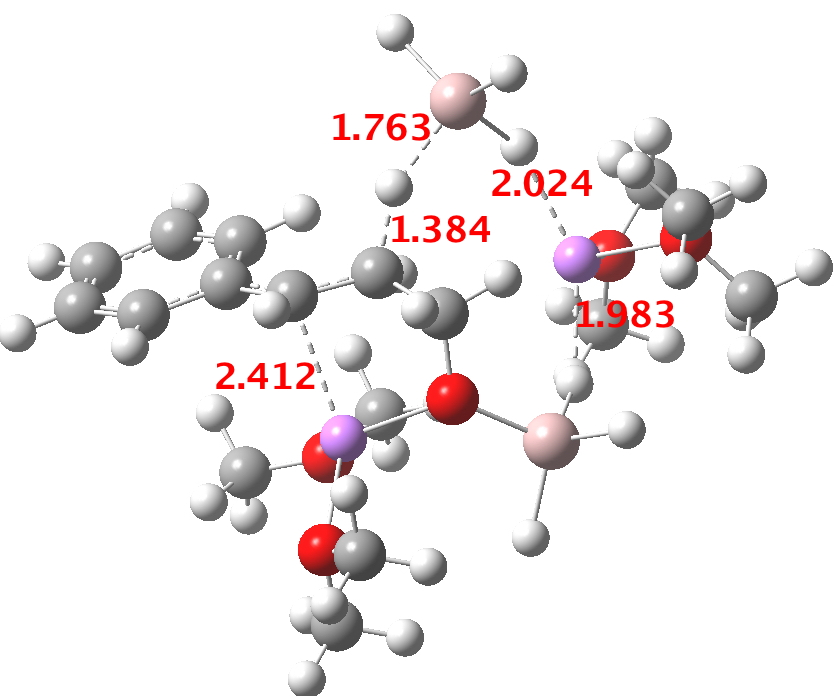
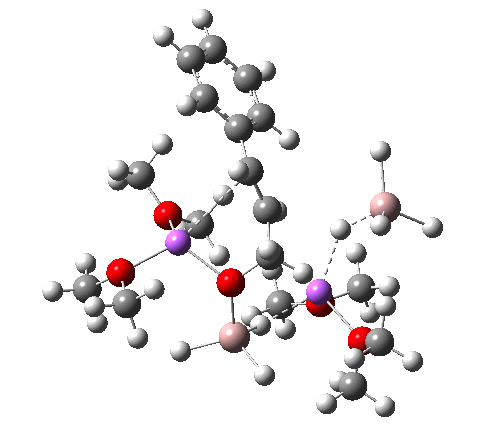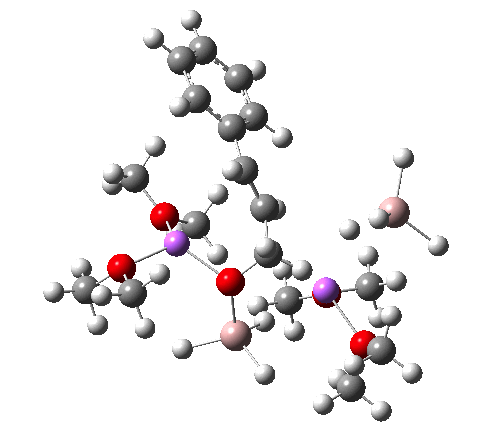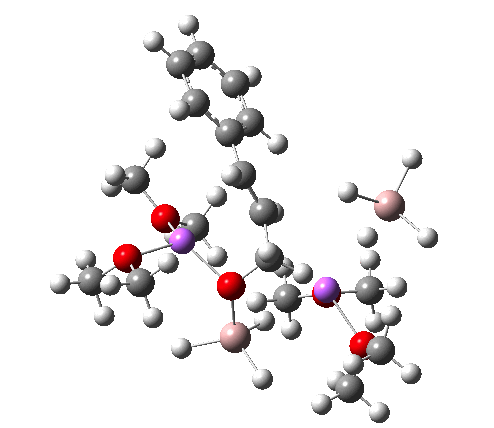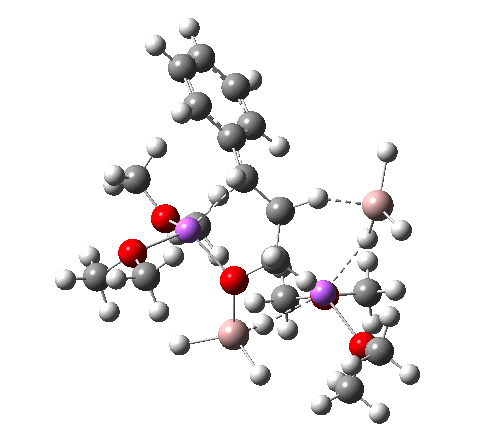
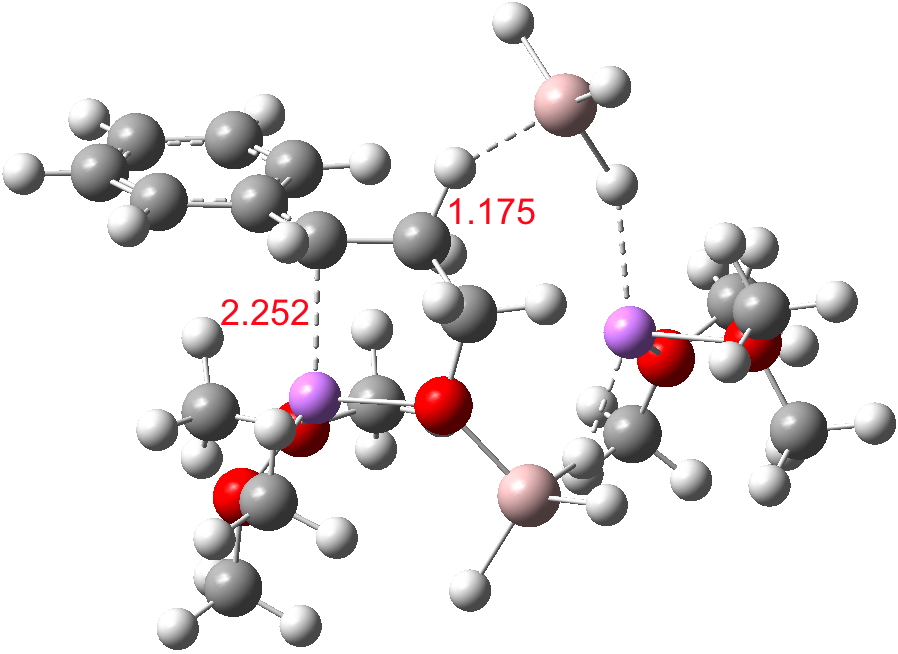
For n=3, we see new solvation patterns, including a dihydrogen bond formed between water and the borohydride at the transition state; ΔG† is 10.0 kcal.mol.
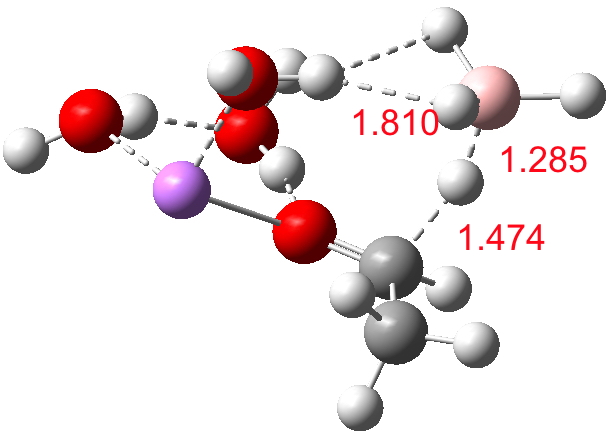
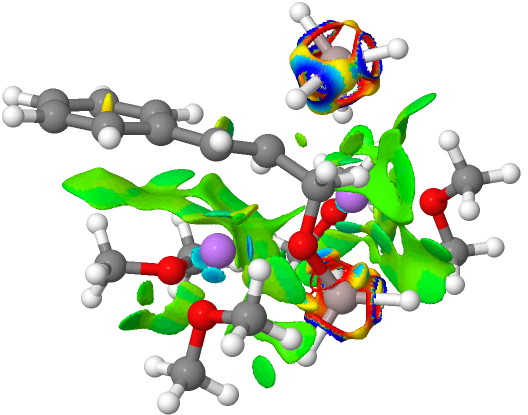
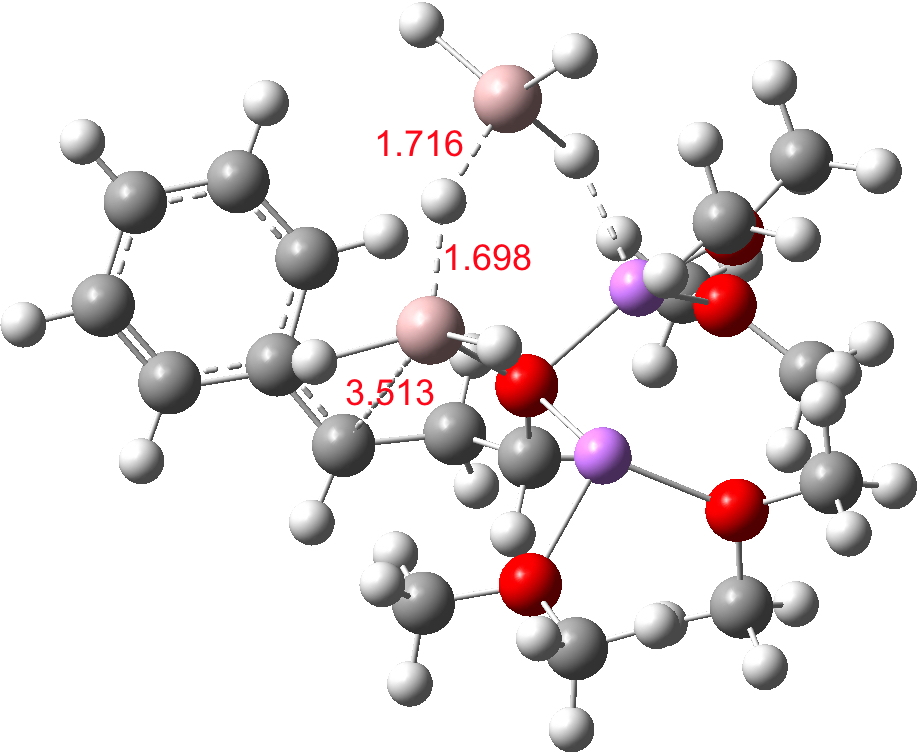
Finally, n=5, where the TS is showing a cage-like structure of complex weak interactions, ΔG† is 11.3 kcal.mol. We see a model where inclusion of explicit solvent molecules can have a significant influence on the size of the barrier obtained.
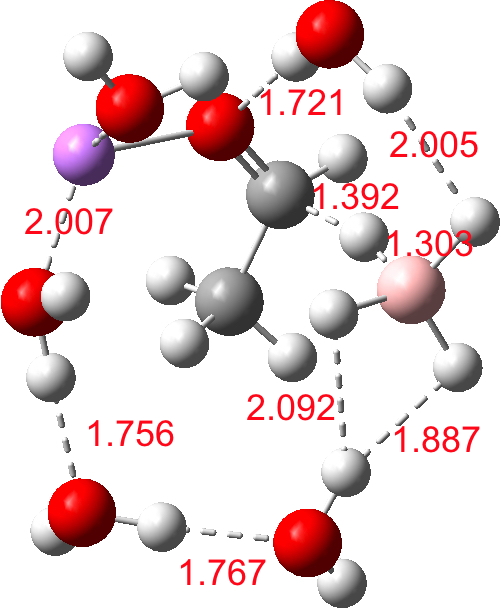
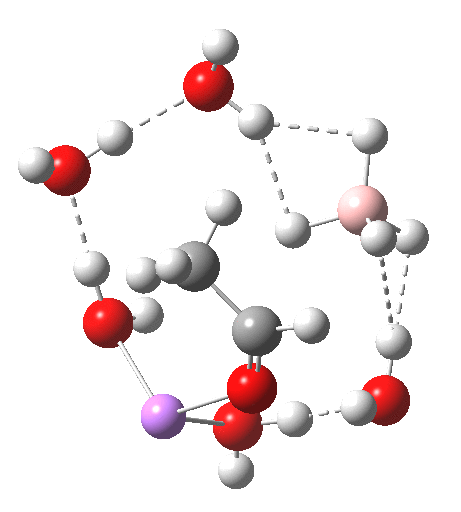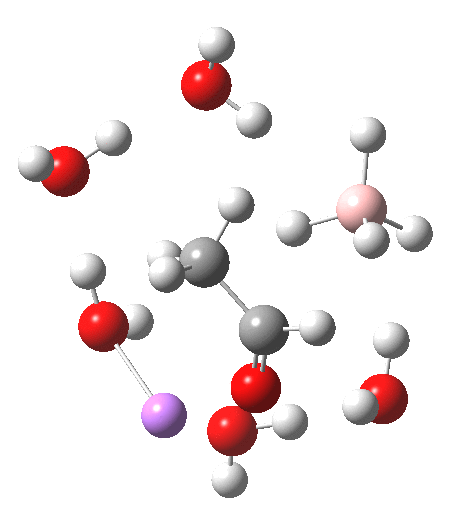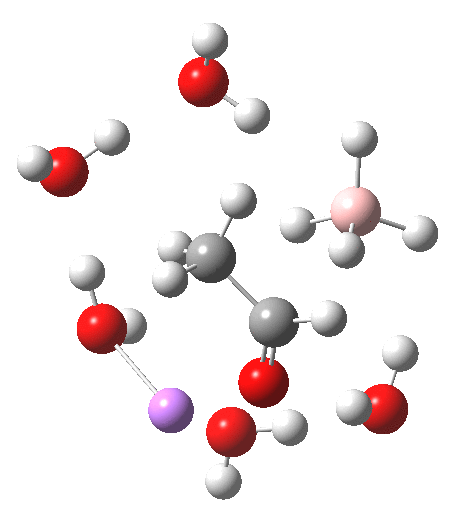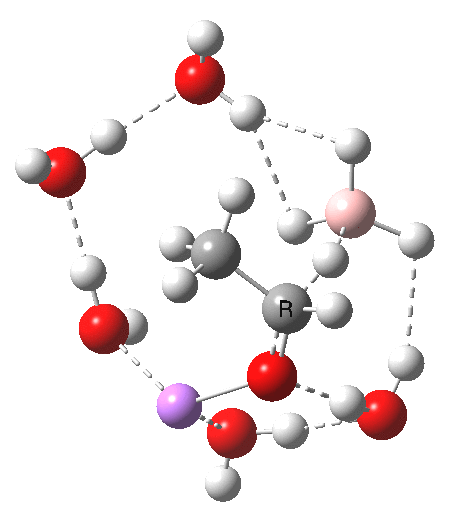
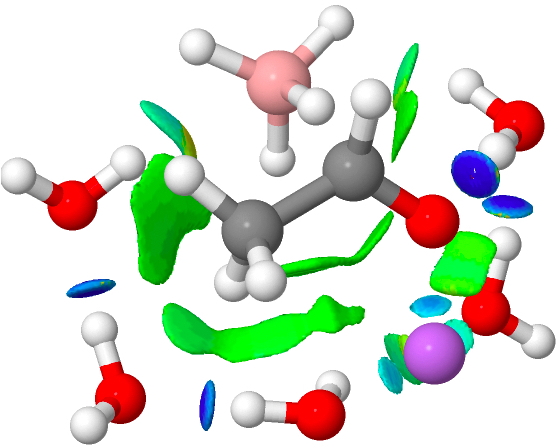
| n | ΔG298‡ | FAIR Data citation |
|---|---|---|
| 2 | 9.6 | [cite]10.14469/ch/191188[/cite] |
| 3 | 10.0 | [cite]10.14469/ch/191189[/cite] |
| 5 | 11.3 | [cite]10.14469/ch/191192[/cite] |
With a mechanistic prototype now identified, it is time to start varying some of the parameters, such as X and R. This will enable us to assess the models built here to see if they reflect reality.








{kind=link}