I am exploring the fascinating diverse facets of a recently published laboratory experiment for undergraduate students.[cite]10.1021/acs.jchemed.7b00566[/cite] Previously I looked at a possible mechanistic route for the reaction between an enal (a conjugated aldehyde-alkene) and benzyl chloride catalysed by base and a chiral amine, followed by the use of NMR coupling constants to assign relative stereochemistries. Here I take a look at some chiroptical techniques which can be used to assign absolute stereochemistries (configurations).
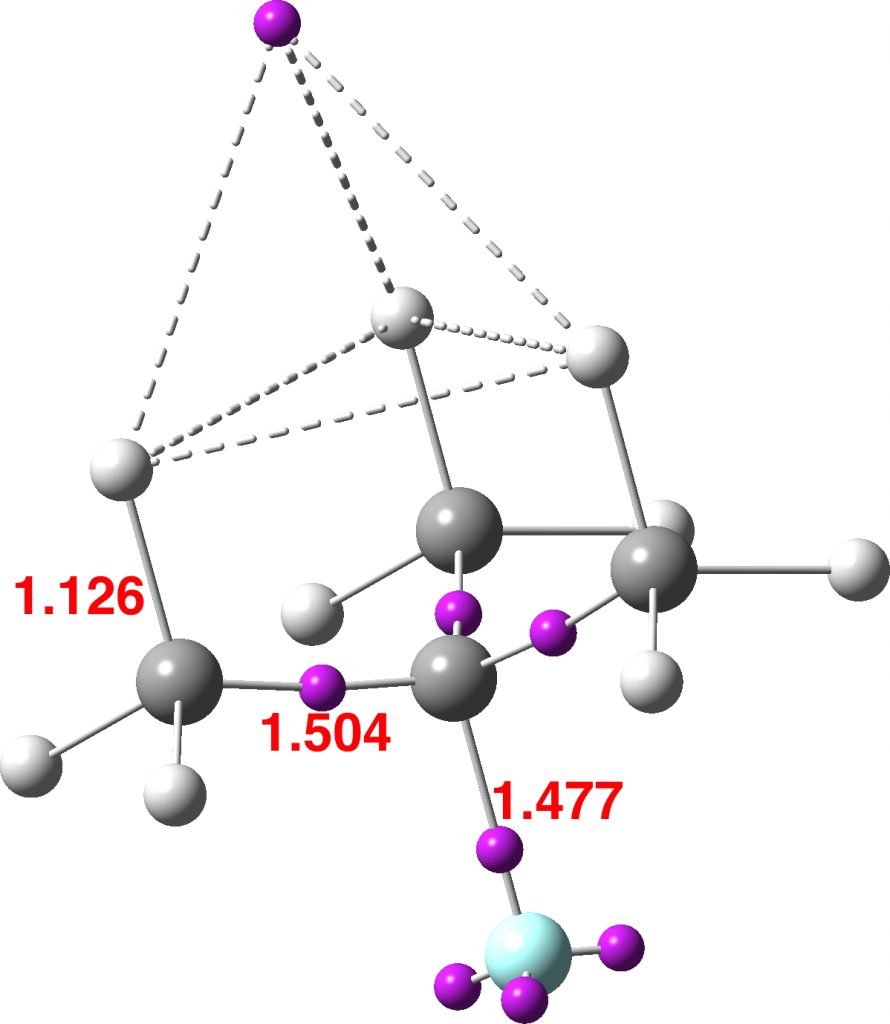
I will focus on the compound 4a, the major stereochemical product of this student laboratory reaction, with the stereochemistry as represented in e.g. the abstract of the main article[cite]10.1021/acs.jchemed.7b00566[/cite] and shown below with added CIP (Cahn-Ingold-Prelog) notation as (1S,2R,3R);‡

Its enantiomer (not shown in the article) is of course;

In the article supporting information[cite]10.1021/acs.jchemed.7b00566[/cite]), the major diasteromer of 4a deriving from use of the S stereoisomer of the prolinol catalyst is reported as having an optical rotation (ORP) [α]D25 of -62.4°, p6 or -58.1°, p5), but the stereo-labels are not added there. On p1 (“based on a student report”) 4a was however labelled as (1R,2S,3S) and the chirality (S) of the catalyst used was also noted in the adjacent experimental procedure. One might then reasonably match (1R,2S,3S)-4a to the S-catalyst and hence (1S,2R,3R)-4a to the R-catalyst. However, in a laboratory environment where both S and R catalysts are in circulation, it is always useful to have procedures available for independent checks.†
There are two methods of assigning absolute chirality, crystallography and chiroptical spectroscopy. The former does require crystalline samples; the latter can use solutions. To cut to the chase, the former method was used for a related compound where the n-heptyl group above is replaced by a p-chlorophenyl substituent (perhaps because the latter imparts suitable crystallinity). On p S123 of the SI of an earlier article[cite]10.1021/acs.joc.5b02801[/cite] the assignment for the p-chlorophenyl derivative was as (1R,2S,3S)-4a for S-catalyst (see DOI: 10.5517/ccdc.csd.cc1mcqg5 OZAXEU). But this procedure is not entirely foolproof; the stereochemistry is decided by interactions between often bulky substituents at the transition state and it might be that here the p-chlorophenyl derivative has different properties from n-heptyl. Moreover bulk solutions may be different in their composition from single crystals. So it is useful to obtain independent proof.
An absolute assignment procedure based on chiroptical methods was first famously used by Kirkwood in 1951 (the Fischer convention is confirmed as a structurally correct representation of absolute configuration).[cite]10.1063/1.1700491[/cite] Such calculations need to take into account e.g. rotational conformers about the two bonds labelled in red above. In the previous post, I had noted variation of up to 2Hz in the calculated 3JHH coupling constants as a result of this mobility. This variation is probably too small to really influence any relative stereochemical interpretations, but is the same true for chiroptical assignments?
In Table 1 we can see whether this is still true for the predicted optical rotation of compound 4a, using two different functionals for the calculation (B3LYP and M062X respectively). The results rather surprised me; a simple bond rotation of an aryl or carbonyl group can invert the sign of the rotation. Clearly the observed optical rotation of -62.4° arises from a suitable combination of different Boltzmann populations of the individual bond rotamers, but to combine these accurately you would need to know the solution populations themselves very accurately and that is quite a challenge. So at this stage, we do not really have totally convincing independent evidence of whether the observed negative optical rotation corresponds to (1S,2R,3R)-4a or to its enantiomer (1R,2S,3S).
|
Table 1. Calculated Optical rotations for (1S,2R,3R)-4a.
FAIR Data DOI: 10.14469/hpc/4678
|
| Conformer |
ORP [α]D, B3LYP+GD3BJ/Def2-TZVPP/SCRF=chloroform
|
ORP [α]D, M062X/Def2-TZVPP/SCRF=chloroform
|
| 4 |
+376 |
+238 |
| 3 |
-335 |
-301 |
| 2 |
-247 |
-223 |
| 1 |
+710 |
+522 |
Next, another chiroptical technique, electronic circular dichroism, or ECD. Here, the sign of the difference in absorption of polarized light (Δε), and known at the Cotton effect, characterises the specific enantiomer. The experimental Cotton effect for compound 4a obtained from S-catalyst (known as 3d in the SI, p S142[cite]10.1021/acs.joc.5b02801[/cite]) can be simply summarised as +ve@315nm and -ve@275nm. Comparison with calculated spectra (Figure S17, p S146-7[cite]10.1021/acs.joc.5b02801[/cite])♣ was performed using a Boltzmann-averaging (albeit based on enthalpies rather than the formally correct free energies), for three significant populations and this procedure matched to (1R,2S,3S). Since the reported calculations were apparently for gas phase (and replacing n-heptyl with methyl) here I have repeated them in the actual solvent used (acetonitrile) and with the heptyl present. Although the ECD responses can still be severely dependent on the conformation, three of the spectra qualitatively agree that the responses at ~300nm♦ and 260 nm are respectively -ve and +ve. This confirms that (1S,2R,3R)-4a is the wrong enantiomer for S-catalyst and that the correct assignment is therefore (1R,2S,3S), as was indeed reported.[cite]10.1021/acs.joc.5b02801[/cite]
|
Table 2. Calculated electronic circular dichroism for
(1S,2R,3R)-4a. FAIR Data DOI: 10.14469/hpc/4678
|
| Conformer |
ECD calculation, ωB97XD/Def2-TZVPP
|
| 4 |
 |
| 3 |
 |
| 2 |
 |
| 1 |
 |
It is still true that the overall the fit between chiroptical experiment and theory can be sensitive to the Boltzmann population, as obtained from e.g. ΔΔG = -RT ln [1]/[2]), where 1 and 2 are two different conformers. ΔΔG is a difficult energy difference to compute accurately. Here is a suggested exercise in the statistics of error propagation. How does an error in ΔΔG propagate to the ratio of concentrations of two conformers [1]/[2]? Or, how accurately must ΔΔG be calculated in order to predict conformer populations to say better than 5%.
One more go at chiroptics, this time Vibrational Circular Dichroism, or VCD. The nature of the chromophore is different, but the principle is the same as ECD. I have deliberately truncated the spectrum to cut off all vibrations below 1000 cm-1 (these being the modes associated with group rotations) but to no avail, the four conformations all still look too different to avoid doing a Boltzmann averaging.
A modern VCD instrument does have one trick up its sleeve for coping with the conformer problem. The sample (as a thin-film) can be annealed down to very low temperatures before the spectrum is recorded. This effectively removes all higher energy forms, leaving just the most stable conformation as the only species present. However, that is an expensive experiment (and instrument!) to use.
There are perhaps some 2 million scalemic molecules (substances where one chiral form is in excess over the mirror image) for which chiroptical properties have been reported, but probably <50,000 crystal structures where absolute configurations have been assigned. Thus the vast majority of absolute configuration assignments have been done either chiroptically or by synthetic correlations (chemical transformations from molecules of known absolute configuration, with the assumption that you know how each transformation affects the chiral centres present). Given some of the difficulties and challenges noted above, it is tempting to conclude that a significant proportion of those 2 million molecules may have been mis-assigned (I once estimated up to 20%). However, we may conclude that the molecules discussed here are safely assigned correctly!
‡No CIP-stereolabels appear in the article itself.[cite]10.1021/acs.jchemed.7b00566[/cite] Perhaps this assignment is omitted in order to provide a student exercise? †There are many errors in stereochemical assignments in the literature. A good many of them may be the result of simple sample mis-labelling.[cite]10.1021/ol901172g[/cite] ♣The caption to Figure S17 states All the simulations are for the 1R,2R,3S absolute configuration. This is probably an error and should read 1R,2S,3S. ♦A correction of ~+15nm is sometimes applied to these values, but not done here.