Deltamethin is a pyrethroid insecticide for control of malaria which has been used for a little while. Perhaps inevitably, mosquitoes are developing resistance to it. So what could be done about countering this? Well, perhaps surprisingly, form a polymorph![1] These crystal structure isomers are often highly undesirable; thus Ritonavir, which changed its polymorphic form during manufacture to become far less active (due it has to be said to insolubility). Now a polymorph of Deltamethin has been discovered, which when applied as a powder, increases its effectiveness more than 10 times against Anopheles mosquitoes and provides a potentially new affordable malaria control solution for countries that are loosing protection.

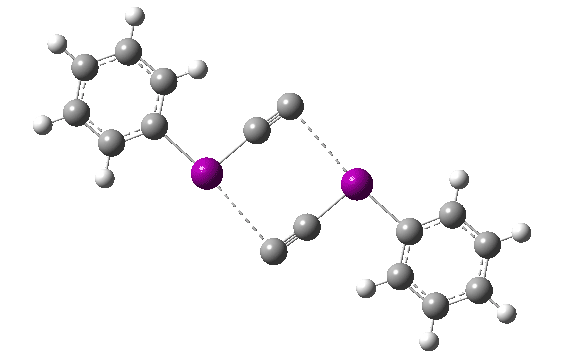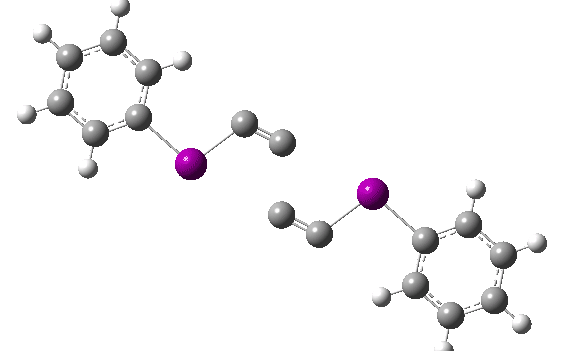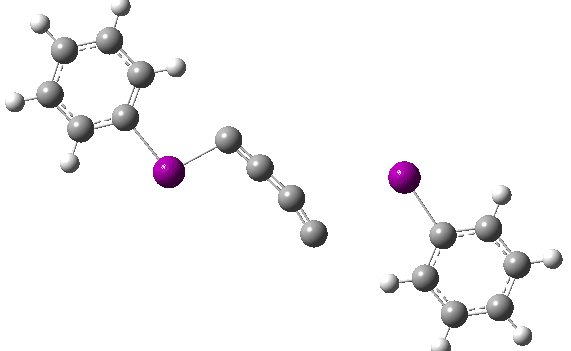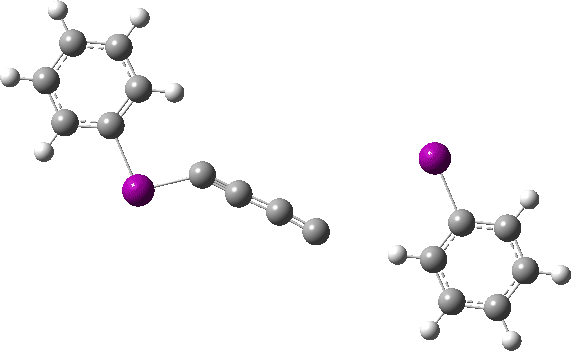
You can view this new polymorph at DOI: 10.5517/ccdc.csd.cc24mrxg and 10.5517/ccdc.csd.cc24mryh. The two structures differ in the torsion angle around the O-C(CN) bond, resulting in differing anomeric interactions between the ester oxygen and the C-CN group.

The anomeric effect can be quantified by the dihedral angle between a donor oxygen lone pair and the accepting (in this case C-CN) bond. The position of the lone pair itself can be localised using the ELF (electron localisation function) method. For one polymorph, this has a torsion angle of 127° and O-C and C-CN lengths of 1.455 and 1.480Å. The other form has an almost orthogonal (i.e. non-interacting) torsion of 79° with lengths of 1.461 and 1.477Å. The first form exhibits some electron pair donation into the C-O bond, shortening it slightly, accompanied by transfer of C-CN bond electrons onto the N, weakening the C-N bond. A very similar effect was noted for a herbicide[2] which resulted in very differing stabilities for two stereoisomers in water.
Just such aqueous hydrolytic stability was indeed reported for Deltamethrin[3] and its other 7 stereoisomers in water (3 stereocentres, 23 = 8). Persistence of any chemical applied to the soil is a very important aspect of its properties; recollect that a now banned insecticide, DDT, suffered from very long soil persistence. The article[3] looks at what happens to Deltamethrin (labelled compound 1) in water in the dark. As it goes, a new isomer labelled 2′ appears and then too decays, all in a period of ~5 days. This new isomer was found to be inactive as an insecticide.

So what is 2′? Well here is a table of all 8 stereoisomers, where 1 is labelled as α-S,1R cis and 2′ is labelled as α-S,1S cis. Here we have only one of the three stereocentres labelled using Cahn-Ingold-Prelog (CIP) notation, the 1-R/S carbon (the convention is to use italics for CIP). The cis designation implies that the 3-position is also S and the cyano carbon remains S. This cis label however conflicts with the abstract to the article, which indicates that the dark water isomerisation is subject to cis/trans isomerisation. So we have a water-induced isomerisation which inverts the configuration of 1R,3R in molecule 1 to apparently 1S,3S in molecule 2′? 
Changes in stereochemistry in Deltamethrin are likely to be the result of forming planar enols and so one might judge the hydrolytic stability by the ease of forming such an enol. Just such a process was the topic of a post on the undesired epimerisation of thalidomide in water. Time for calculations (FAIR data doi: 10.14469/hpc/8020 ) at the B3LYP+GD3+BJ/Def2-TZVPP/SCRF=water computational level. Firstly, the free energy of the enol form on the cyclopropane by relocation of the proton at C-1 is 32.0 kcal/mol higher. However, forming the enol by relocating the proton on the carbon bearing the CN group to the nitrogen is only 20.0 kcal/mol higher in energy. With a small further barrier for the TS for proton removal expected, this latter enol is within an accessible energy range for room temperature reaction, whereas the previous enol is not. So the hypothesis is that 2′ is actually the stereoisomer labelled 2 in Table 1, ie R,R,R and not S,S,S. The free energy of this R,R,R diastereoisomer is calculated 1.1 kcal/mol higher than 1 itself, which would represent about 13% of this isomer at equilibrium.
I have shown here how uncertainty caused by how to reconcile the stereochemistry of eg Figure 4 and Table 1[4] in modern terms can perhaps be lessened by performing “reality check” calculations on possible multiple interpretations to reduce them to the most probable. We also find that a relatively old and much used bioactive compound can have surprises lurking around the corner, in this case by a simple recrystallisation that results in a new form being discovered without having to do any other chemistry. Still, how much longer will pyrethroid insecticides be used?
References
- J. Yang, B. Erriah, C.T. Hu, E. Reiter, X. Zhu, V. López-Mejías, I.P. Carmona-Sepúlveda, M.D. Ward, and B. Kahr, "A deltamethrin crystal polymorph for more effective malaria control", Proceedings of the National Academy of Sciences, vol. 117, pp. 26633-26638, 2020. https://doi.org/10.1073/pnas.2013390117
- P. Camilleri, D. Munro, K. Weaver, D.J. Williams, H.S. Rzepa, and A.M.Z. Slawin, "Isoxazolinyldioxepins. Part 1. Structure–reactivity studies of the hydrolysis of oxazolinyldioxepin derivatives", J. Chem. Soc., Perkin Trans. 2, pp. 1265-1269, 1989. https://doi.org/10.1039/p29890001265
- R.J. Maguire, "Chemical and photochemical isomerization of deltamethrin", Journal of Agricultural and Food Chemistry, vol. 38, pp. 1613-1617, 1990. https://doi.org/10.1021/jf00097a039
















































{kind=link}