October 9th, 2014
This second report highlights two “themes”, or common ideas that seem to emerge spontaneously from diversely different talks. Most conferences do have them.
The first is “embedding“, which in this context means treating different parts of a probably complex molecular system at different levels of theory. Thus Emily Carter in her plenary described how a periodic crystal treated by density functional theory, or DFT could have an embedded component in which the electronic structures are described instead by multi-reference correlated wave functions (CAS-PT2). She illustrated this by discussing what happens when a triplet state oxygen molecule approaches the surface of an aluminium crystal, and (mostly) dissociates into surface bound oxygen atoms with Al-O bonds. The spin state of the oxygen changes smoothly to an overall singlet, with a rapid transfer of charge at the saddle point in the potential energy surface. The numbered of embedded Al atoms had to be at least a cluster of 14 to reproduce the observed reaction barriers (DFT on its own gets a zero barrier!). This sort of study is important in understanding the details of what is happening in metal surface catalysis.
Arieh Warshel then addressed the same theme with his own talk entitled Multiscale Modeling of Complex Biological Systems and Processes. Here you got quantum embedding in a mechanical force field description of some very large molecules. This was a broad brush talk, but what I did get out of it was the concept of asymmetry in molecular systems. Whereas an organic chemist thinks of asymmetry as often relating to just a single chiral carbon centre in a molecule, nature operates on vaster scales. Thus the enzyme ATPase has a molecular axle or spindle, which rotates to assemble the phosphate groups one at a time. This spindle rotates asymmetrically, i.e. always in a specific direction, and Warshel attempts to describe the origins of this rotational asymmetry at a molecular level. Well, this is Nobel prize winning stuff! He followed this up with filaments that “walk” along surfaces in one (asymmetric) direction, first lifting up one point of attachment, and then re-attaching at a different point such that the filament develops a clear sense of direction in its walk. This of course is all done with molecular dynamics, and (I think) has its origins in subtle electrostatics.
Stefan Grimme in his plenary also described dynamic processes, this time those that happen in a mass spectrometer when a molecule is ionised by electron impact. Removal of an electron produces a complex set of ionised states, in which many different single bonds may be weakened due to this ionisation. He developed simplified DFT (sDFT) methods that can be applied to molecular dynamics, and assembled a “black box” which predicts the expected fragmentations over a time scale of a ps or so. By sampling the trajectories, he estimated the intensities of the various positively charged species and overlaid this on the observed EI-MS. The agreement was often spectacular. A particularly interesting example was the fragmentation of taxol. Here, no molecular ion is found, only much lighter ions. The molecular dynamics shows that rather than consecutive single-bond fragmentations, you instead get multiple bonds more or less all fragmenting at the same time. Tougher was to reproduce rearrangements, such as the McLafferty. Here, the semi-empirical method OM2 was more successful. His work means you can just “dial-a-mass-spectrum” and he speculates whether getting a good fit with the observed spectrum could tell you subtle aspects of the gas-phase molecular species, what its tautomeric state might be or perhaps even its conformation. He also described large-scale (800+) atom simulations of electronic circular dichroism (ECD) spectra of organometallic systems. Octahedral complexes can be prepared in chiral form, and this theoretical ECD treatment allows determination of absolute configuration of these often non-crystalline systems. Here you often need to compute 1000 or more electronic states, and if you have ever tried such ECD simulations, you will know that this is a lot of states!
We had been expecting Stefan to talk about dispersion effects in molecules, another emerging theme. Instead lots of other people mentioned them. In my talk I showed how including a D3-dispersion correction could dramatically change the predicted enantioselectivity of a chiral aldol condensation.[1]
The above observations of course cannot be in the least representative; typical of a modern conference there are five parallel sessions and 400+ posters, and so it represents a highly personal and selective snapshot.
References
Tags: Complex Biological Systems, condensation, gas-phase molecular species, metal surface catalysis, molecular systems, non-crystalline systems, organic chemist, organometallic systems, potential energy surface, representative, Stefan Grimme, Thus Emily Carter
Posted in Interesting chemistry, WATOC reports | 1 Comment »
October 6th, 2014
I am attending a conference. Plenaries at such events can sometimes provide interesting pointers on things to come (and sometimes they simply point to things past). At WATOC2014 in Santiago Chile, the first plenary was by Paul Ayers with the impressive title “Concepts for organising chemical knowledge” which certainly sounds as if it is pointing forward!
WATOC stands for World Association of Theoretical and cOmputational Chemists (yes, the acronym does not expand) and so the organising concepts of course relate to quantum mechanics. Ayer’s talk however was more about chemical philosophy, and how we may continue to organise chemical rules according to sound quantum mechanical principles. He bases these principles on observables, rather than just mathematical formulations. The start point is electron distributions (I have noted elsewhere that these are indeed measurable observables, although the crystallography required to measure bond-based electron density is only rarely carried out) in the form of conceptual DFT. He strives to find examples that cannot be explained by e.g. the older ideas of molecular orbitals (as obtained from solution of a single reference determinant) and in particular to avoid the modern syndrome that as we improve our ability to achieve numerical accuracy, we tend to lose our insight into the phenomenon being studied. One example he gives is that of alkynes such as FC≡CF. If one removes a π-electron from the molecule, this should result in a decrease in the electron density distribution? Wrong. It actually increases the σ-electron density in the nodal planes of the lost π-electron. Ayer’s point is that Conceptual DFT provides a simple framework for explaining such observations, unlike MO theory.
But the most interesting moment came with discussion of Bader’s QTAIM methods (often used in this blog), as used by Ayers to provide a formulation of an electronic stress tensor approach to molecules. Expressed as Ehrenfest Forces and the corresponding Hessian, these provides a handle on the forces on bonds, or in simpler language, telling us “how bonds might move“. Think of it as the electronic equivalent of how nuclei move in molecules (aka vibrations). The reason my ears pricked up is that I have often discussed how organic chemists tend to conceptualize “how bonds might move” by using curly arrow pushing. This concept, dating back to Robinson’s original example is often dismissed by theoreticians (and it has to be said by some organic chemists) as having little sound theoretical foundation; it is pure symbolism, and not to be over-interpreted. So perhaps electronic stress tensors based on Ehrenfest Forces might indeed tell us whether the hugely successful curly arrow pushing formalism (itself based on Lewis’ ideas of the shared electron bond formulated in 1916) might yet receive the theoretical makeover that it badly needs.
But before I end, one more example given by Ayers; keto-enol tautomerism in say propanone. Here a C-H bond breaks and an OH bond forms. The Ehrenfest Hessian indeed tells is that this is indeed “how the bonds want to move“. At the point the “audience” (in the form of a question) injected that sense of excitement that a conference occasionally provides. According to MO theory (!), the [1,3] sigmatropic shift of a hydrogen atom is a forbidden pericyclic process! How might the conceptual DFT electronic stress tensor approach tell us this? I have to confess I am not sure I understood Ayers’ answer at this point. Perhaps more work is needed before MOs are discarded entirely.
Oh, one more comment. Conference talks, presented in the form of slides which might linger on the projected screen for only a few seconds, rarely allow the audience to take much more than cursory notes. In particular, without some more research, I cannot here cite any of the references to Ayer’s recent work, since I did not have time to make a note of them.
Postscript: An article by Paul Geerlings, Paul Ayers et al on the topic of the Woodward-Hoffmann rules has in fact been published[1].
Postscript2: I have computed the density difference isosurface for FCCF as noted above. It is shown as a thumbnail below (blue is decreased density on forming the cation, red is the increased density). Click on this to rotate.

Click for 3D
References
- P. Geerlings, P.W. Ayers, A. Toro-Labbé, P.K. Chattaraj, and F. De Proft, "The Woodward–Hoffmann Rules Reinterpreted by Conceptual Density Functional Theory", Accounts of Chemical Research, vol. 45, pp. 683-695, 2012. https://doi.org/10.1021/ar200192t
Tags: chemical knowledge, chemical philosophy, chemical rules, Chile, Paul Ayers, Paul Geerlings, Santiago, World Association of Theoretical and cOmputational Chemists
Posted in Interesting chemistry | No Comments »
September 16th, 2014
ELNs (electronic laboratory notebooks) have been around for a long time in chemistry, largely of course due to the needs of the pharmaceutical industries. We did our first extensive evaluation probably at least 15 years ago, and nowadays there are many on the commercial market, with a few more coming from opensource communities. Here I thought I would bring to your attention the potential of an interesting new entrant from the open community.
My very first post on this blog six years ago related to incorporation of the Jmol molecular viewer into posts, and it has been a feature of many since. A little more than two years ago, Jmol was recast into JSmol. This had become possible because JavaScript engines built into modern web browsers were finally getting the sort of performance needed to display molecules (years and years ago, lets say ~1990, such display required very fancy hardware kit such as Silicon Graphics workstations). Around the same time, another well-established Java-based molecule sketcher, JME (Java molecular editor) also became JavaScript based. My own interest in this sort of Web-based behaviour actually crystallised last December, when I decided to refactor my own lecture notes into a tablet-friendly format using JSmol, with some questions directed at the formidably excellent Jmol discussion list. One of these related to how students might annotate such lecture notes with chemical sketches and store the results for future study or revision. Otis Rothenberger starting exploring various mechanisms for such local storage (using Web browsers), and in the last month or so has found a way of exploiting something called HTML5 local storage, which allows the sort of capacity needed. These three technologies have now come together on Otis’ site, which you can now view as CheMagic Notebook (this might be a .com site, but I believe the concept is very much open).
 Together with the Virtual model kit (VMK, itself now part of JSmol) this combination is starting to resemble a very interesting mechanism for creating an immersive lecture note environment, almost you might say a lecture note ecosystem. I would argue that for the first 30 years of the digital document era, most people preparing lecture notes became mesmerised (distracted?) by the need to print the outcomes with complete fidelity. It is only recently that the focus has turned to “beyond the PDF” (or beyond the PPT) and much richer mechanisms. So now we have lecture notes morphing into an ecosystem where:
Together with the Virtual model kit (VMK, itself now part of JSmol) this combination is starting to resemble a very interesting mechanism for creating an immersive lecture note environment, almost you might say a lecture note ecosystem. I would argue that for the first 30 years of the digital document era, most people preparing lecture notes became mesmerised (distracted?) by the need to print the outcomes with complete fidelity. It is only recently that the focus has turned to “beyond the PDF” (or beyond the PPT) and much richer mechanisms. So now we have lecture notes morphing into an ecosystem where:
- the objects themselves can be interactive (3D models, spectra, animations etc)
- or reference further models and associated data held in digital repositories
- or built from scratch in response to stimulation from peers, tutorials, workshops or lectures (using eg VMK or JME)
- and such annotations in effect themselves can be spliced into the student’s own copy of these notes,
- with the whole being regarded as a running notebook created from the initial seed of a lecturer’s materials augmented by the student’s own annotations.
I have focused here on where I started, i.e. refactoring my own lecture notes. But the above concepts could easily morph into eg a research project notebook, a rebundling into smaller segments which are themselves published into digital repositories (and there assigned their own persistent digital object identifiers) and ultimately further morphing into scholarly articles submitted to say a journal. These could represent a continuum, not discrete (and non-communicating) objects.
So will “lecture notes” actually start to change from their conventional (printable) form into something related to the above? Well, I have not addressed the largest hurdle preventing this; giving the content creators (i.e. the lecturers) the training, skills and most importantly the motivation to start to venture down this pathway. Otis has shown it should be technically possible. Come back and revisit this post in ten years time to see what actually did happen!
Tags: .com, chemical sketches, Java, JavaScript, lecturer, molecular editor, PDF, pharmaceutical industries, Silicon Graphics, three technologies, web browsers, Web-based behaviour
Posted in Chemical IT | No Comments »
September 8th, 2014
In the beginning (taken here as prior to ~1980) libraries held five-year printed consolidated indices of molecules, organised by formula or name (Chemical abstracts). This could occupy about 2m of shelf space for each five years. And an equivalent set of printed volumes from the Beilstein collection. Those of us who needed to track down information about molecules prior to ~1980 spent many an afternoon (or indeed a whole day) in the libraries thumbing through these weighty volumes. Fast forward to the present, when (closed) commercial databases such as SciFinder, Reaxys and CCDC offer information online for around 100 million molecules (CAS indicates it has 89,506,154 today for example). These have been joined by many open databases (e.g. PubChem). All these sources of molecular information have their own way of accessing individual entries, and the wonderful program Jmol (nowadays JSmol) has several of these custom interfaces programmed in. Here I describe some work we have recently done[1] on how one might generalise access to an individual molecule held in what is now called a digital data repository.
Such repositories are gradually becoming more common. Unlike most (all?) of the bespoke molecular repositories noted above, metadata (XML) resourcemap standards have been developed[2] for data repositories to enable rich and open searches and to help in the discoverability of individual entries (e.g. OAI-ORE). Each dataset is characterised by a DOI (digital object identifier), just like individual articles found in a conventional journal. However, there is an issue in quoting just a conventional DOI to describe a dataset. The DOI points to what is called the article landing page in the journal. A landing page which by and large is meant to be navigated by a human. To get a flavour for how this works (or more accurately does not work) for data, visit this DOI[3] for an entry in the CCDC crystal database noted above (and about which I have previously blogged). In essence, a human is needed to complete the requested information in order to proceed to retrieving the data. Data, I contend here, should not need a landing page. It can benefit from being passed straight on to e.g. a visualising program such as JSmol. So a mechanism is needed to encapsulate any bespoke (and potentially changeable) access path to the data by expressing it instead in standard metadata form.
In our first solution to this issue, and the one illustrated here, we used a standard known as 10320/loc[2]. A datafile need only be specified by its DOI (or more generically, its handle) to be recovered from the data repository; no landing page need be involved (and no human need ponder what next to do with the data).
- First, let me reference a molecule (as it happens the one described in the preceding post), using the normal invocation[4]. This will take you to a conventional landing page.
- The next example is the same dataset, but this time with the landing page replaced by a Javascript/JSmol wrapping. This is achieved using a utility which is itself packaged up and placed on a repository (shortdoi: vjj)[5], and which is embedded here for you to try out. If you want the technical detail, read about it here.[1]
There is more to come. But you will have to wait for part 2!
References
- M.J. Harvey, N.J. Mason, and H.S. Rzepa, "Digital Data Repositories in Chemistry and Their Integration with Journals and Electronic Notebooks", Journal of Chemical Information and Modeling, vol. 54, pp. 2627-2635, 2014. https://doi.org/10.1021/ci500302p
- "DOI Name 10320/loc Values"http://doi.org/10320/loc
- Jana, Anukul., Omlor, Isabell., Huch, Volker., Rzepa, Henry S.., and Scheschkewitz, David., "CCDC 967887: Experimental Crystal Structure Determination", 2014. https://doi.org/10.5517/cc11h55w
- H.S. Rzepa, N. Mason, and M J Harvey., "Retrieval and display of Gaussian log files from a digital repository", 2014. https://doi.org/10.6084/m9.figshare.1164282
Tags: Digital Object Identifier, XML
Posted in Chemical IT | No Comments »
September 6th, 2014
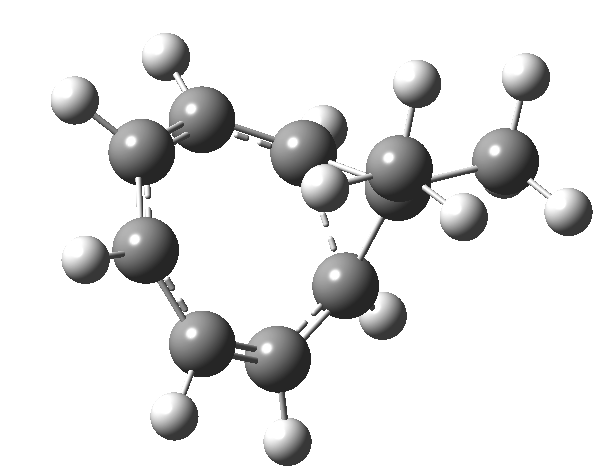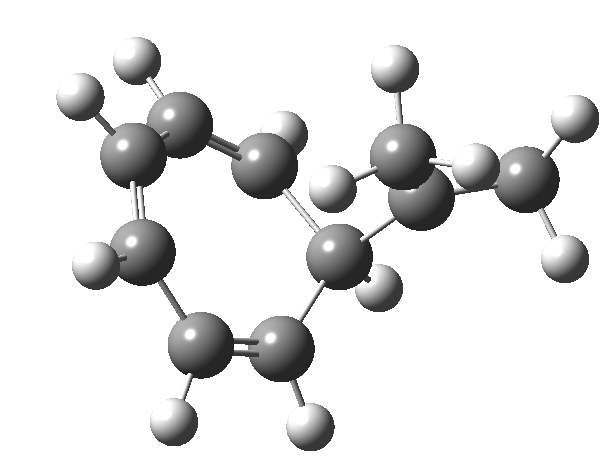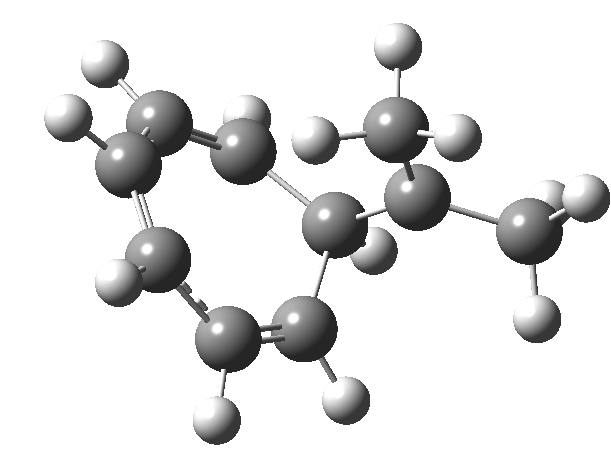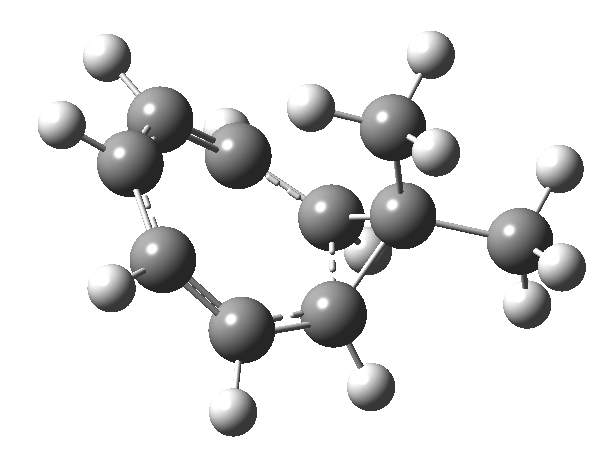
In the previous posts, I explored reactions which can be flipped between two potential (stereochemical) outcomes. This triggered a memory from Alex, who pointed out this article from 1999[1] in which the nitrosonium cation as an electrophile can have two outcomes A or B when interacting with the electron-rich 2,3-dimethyl-2-butene.  NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1]
NMR evidence clearly pointed to the π-complex A as being formed, and not the cyclic nitrosonium species B (X=Al4–). If you are wondering where you have seen an analogy for the latter, it would be the species formed when bromine reacts with an alkene (≡ Br+, X=Br– or Br3–). The two structures are shown below[1]  Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest.
Since the topic that sparked this concerned pericyclic reactions, it seemed possible that if it had been formed, species B would immediately undergo a pericyclic electrocyclic reaction to form the rather odd-looking cation C, which might then be trapped by eg X(-) to form the nitrone D. So this post is an exploration of what happens when X-NO (X= CF3COO, trifluoracetate) interacts with 2,3-dimethyl-2-butene, as an illustration of what can be achieved nowadays from about 2 days worth of dry-lab computation as a prelude to e.g. an experiment in the wet-lab (it would take a little more than two days to achieve the latter I suspect). Hence computationally directed synthesis. The model is set up as ωB97XD/6-311G(d,p)/SCRF=chloroform. A transition state is located[2] and the resulting IRC (below) [3] does not quite have the outcome the above scheme would suggest. 

 Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.
Neither A nor B is formed; instead it is the tetrahedral species E, which is ~15 kcal/mol endothermic.  I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate).
I should immediately point out that this is not inconsistent with the formation of A as previously characterised[1]. That is because this experiment was conducted with a non-nucleophilic counter-anion (X=Al4–), whereas in the computational simulation above, we have a nucleophilic anion (X= CF3CO2–). What a difference the inclusion of a counter-ion in the calculation can have! The barrier however (~35 kcal/mol) is a little too high for a facile thermal reaction. In the second of this two-stage reaction, E now ring-opens to form the anticipated D[4] with quite a small barrier of ~6 kcal/mol, but a highly exothermic outcome. I ask this question about it; can this still be described as a pericyclic process? (there is some analogy to the electrocyclic ring opening of a cyclopropyl tosylate). 
 So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
So what are the conclusions? Well, because of the rather high initial barrier, the alkene will need activation (by electron donating substituents, perhaps OMe) for the reaction to become more viable. But if it works, it could be an interesting synthesis of nitrones (I have not yet searched to find out if the reaction is actually known).
References
- G.I. Borodkin, I.R. Elanov, A.M. Genaev, M.M. Shakirov, and V.G. Shubin, "Interaction in olefin–NO+ complexes: structure and dynamics of the NO+–2,3-dimethyl-2-butene complex", Mendeleev Communications, vol. 9, pp. 83-84, 1999. https://doi.org/10.1070/mc1999v009n02abeh000995
- H.S. Rzepa, "C8H12F3NO3", 2014. https://doi.org/10.14469/ch/24979
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162797
- H.S. Rzepa, "Gaussian Job Archive for C8H12F3NO3", 2014. https://doi.org/10.6084/m9.figshare.1162676
Tags: CF 3, CF 3 CO, COO, simulation
Posted in pericyclic, reaction mechanism | 2 Comments »
August 18th, 2014
This post, the fifth in the series, comes full circle. I started off by speculating how to invert the stereochemical outcome of an electrocyclic reaction by inverting a bond polarity. This led to finding transition states for BOTH outcomes with suitable substitution, and then seeking other examples. Migration in homotropylium cation was one such, with the “allowed/retention” transition state proving a (little) lower in activation energy than the “forbidden/inversion” path. Here, I show that with two electrons less, the stereochemical route indeed inverts. First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1]
First, a [1,4] alkyl shift with inversion at the migrating carbon (ωB97XD/6-311G(d,p)/SCRF=chloroform); as a four-electron process, this is the “allowed” route.[1]  The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2]
The “forbidden” route corresponds to retention of configuration at the migrating carbon.[2]  The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post.
The barriers for each process can be seen below from the IRCs. That for inversion is ~4.5 kcal/mol lower than retention. This nicely transposes the values for the six-electron homologue shown in the previous post. 
 There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
There is one more nugget of insight that can be extracted. The start/end-point for the six-electron process (homotropylium cation) was, as the name implies, homoaromatic. Now, with a four-electron system we also have an inverse. Nominally, we should now end with homo-antiaromaticity (but see [3]). But antiaromaticity is avoided whenever possible, and so the homoaromatic bond observed in homotropylium is not formed. It resolutely remains a σ-bond (1.48Å) thus sequestering two electrons, and the remaining two electrons simply form a delocalised allyl cation. With the six-electron homotropylium, reactant/product were stabilised by that additional (homo)aromaticity, thus inducing a relatively high barrier. With the four-electron system here, no such reactant/product stabilisation occurs, and hence the reaction barriers are now significantly lower. A rather neat pedagogic example.
References
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142175
- H.S. Rzepa, "Gaussian Job Archive for C8H11(1+)", 2014. https://doi.org/10.6084/m9.figshare.1142174
- C.S.M. Allan, and H.S. Rzepa, "Chiral Aromaticities. A Topological Exploration of Möbius Homoaromaticity", Journal of Chemical Theory and Computation, vol. 4, pp. 1841-1848, 2008. https://doi.org/10.1021/ct8001915
Tags: activation energy, reactant/product
Posted in pericyclic, reaction mechanism | 2 Comments »
August 12th, 2014
One thing leads to another. Thus in the previous post, I described a thermal pericyclic reaction that appears to exhibit two transition states resulting in two different stereochemical outcomes. I noted that another such reaction appeared to be a [1,6] carousel migration in homotropylium cation,[1] where transition states for both retention and inversion of the configuration of the migrating group (respectively formally allowed and forbidden) were reported (scheme below). Here I explore this system further.  Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array.
Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array. 

 The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
| HONBO (two) |
HONBO-2 |
 Click for 3D |
 Click for 3D |
At the transition state for the [1,6] migration, the same NICS probe registers a value of +2.6 ppm[4], now firmly in the non-aromatic zone. So this reaction is characterised by two zones, ring-aromatic ones at the start and the end of the process, and a higher energy non-aromatic one in the middle of the reaction pathway as ~enclosed by the region of IRC ± 4.2. The homo-bond in the aromatic zone starts with a length of 1.74Å, reduces to 1.53Å at the transition state and ends up as a normal aromatic bond of length 1.41Å. Meanwhile, the relocated homo-bond changes in the opposite sense, starting as a normal aromatic length of 1.41Å, becoming 1.53Å at the transition state and ending as a homo-length of 1.74Å. Presumably, virtually full strength homoaromaticity can be sustained for a homo-bond of 1.74Å, but as that bond mutates to a σ-bond of 1.53Å, the cyclic conjugation falls off the edge of the cliff, to be restored only at the end. Pericyclic reactions are themselves said to sustain aromatic transition states,[5] and so a simplistic way of looking at this is that the “aromaticity” relocates (or morphs) from the reactant to the transition state, and then back again during the course of the migration. A reaction path from which one can indeed learn a lot.
Now to the pathway in which the migrating group retains configuration. This is no longer a single step concerted reaction,[6] since at the half-way point we no longer have a transition state but a shallow intermediate (~IRC +2, [7]). It (formally at least) becomes a two-step non-concerted process, and the overall barrier is ~5 kcal/mol lower than for the inversion path. The aromaticity changes in a similar manner to before (i.e. IRC ~-5).


So this emerges as not quite the example I thought it was, but nonetheless unusual with the “forbidden” pathway being concerted and the “allowed” pathway being non-concerted. Molecular dynamics on these two systems would indeed be interesting to see what proportion of the trajectories go via each pathway.
References
- A.M. Genaev, G.E. Sal’nikov, and V.G. Shubin, "Energy barriers to carousel rearrangements of carbocations: Quantum-chemical calculations vs. experiment", Russian Journal of Organic Chemistry, vol. 43, pp. 1134-1138, 2007. https://doi.org/10.1134/s1070428007080076
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134556
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135694
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135695
- H.S. Rzepa, "The Aromaticity of Pericyclic Reaction Transition States", Journal of Chemical Education, vol. 84, pp. 1535, 2007. https://doi.org/10.1021/ed084p1535
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135668
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134559
Tags: chemical shift, higher energy, Sangean Table Top Portable Audio Device
Posted in pericyclic, reaction mechanism | 5 Comments »
August 10th, 2014
In my first post on the topic, I discussed how inverting the polarity of the C-X bond from X=O to X=Be (scheme below) could flip the stereochemical course of the electrocyclic pericyclic reaction of a divinyl system. This was followed up by exploring what happens at the half way stage, i.e. X=CH2, the answer being that one gets an antarafacial pathway as with X=O. Here I fill in another gap, X=BH to see if a metaphorical microscope can be used to view the actual region of the “flip” to a suprafacial mode. This time, uniquely, it proved possible to locate TWO transition states for this process, one suprafacial[1] and one antarafacial[2], this latter being 10.5 kcal/mol lower in ΔG† (ωB97XD/6-311G(d,p)/SCRF=dichloromethane). It is quite rare to be able to find BOTH stereochemical outcomes of a thermal pericyclic reaction.‡
This time, uniquely, it proved possible to locate TWO transition states for this process, one suprafacial[1] and one antarafacial[2], this latter being 10.5 kcal/mol lower in ΔG† (ωB97XD/6-311G(d,p)/SCRF=dichloromethane). It is quite rare to be able to find BOTH stereochemical outcomes of a thermal pericyclic reaction.‡
First, the antarafacial IRC (X=BH)[3]. There are several interesting features. Note at IRC = -8, the divinyl compound appears as a Hidden Intermediate (HI), having formed from a compound where the HB=C substituent has ring opened from a cyclobutene-like precursor (initial electrocyclic). If you watch the animation, you can see the antarafacial bond forming from the bottom face of the vinyl group on the left to the top face of the vinyl group in the HI on the right (antarafacial=conrotation). Because the entire process is concerted (no real intermediates participate), we have here an unusual pericyclic cascade where one electrocyclic reaction is immediately followed by another quite different one.

 Now for the suprafacial IRC[4]. It is pretty similar to the previous path, but again if you inspect very carefully you will see that it is the TOP face of the vinyl group on the left forming the bond to the TOP face of the vinyl group on the right (suprafacial/disrotation).
Now for the suprafacial IRC[4]. It is pretty similar to the previous path, but again if you inspect very carefully you will see that it is the TOP face of the vinyl group on the left forming the bond to the TOP face of the vinyl group on the right (suprafacial/disrotation). 

 You might ask if the molecules used here are realistic, i.e. could they form the basis of real reactions to be conducted in a laboratory? Well, the C=B-C fragment has 9 hits in the CCDC crystal database (none for C=B-H). One example is cited here.[5]. So, yes, possibly a realistic system, except the barriers do look too high. Perhaps suitable substituents might help? But even if this could not be carried out in a test-tube, it does teach one about pericyclic reactions and how one might manipulate them.
You might ask if the molecules used here are realistic, i.e. could they form the basis of real reactions to be conducted in a laboratory? Well, the C=B-C fragment has 9 hits in the CCDC crystal database (none for C=B-H). One example is cited here.[5]. So, yes, possibly a realistic system, except the barriers do look too high. Perhaps suitable substituents might help? But even if this could not be carried out in a test-tube, it does teach one about pericyclic reactions and how one might manipulate them.
‡One such is the [1,6] sigmatropic shift in homotropylium cation involving migration of a Me2C+ group, where the “allowed” process in which the migrating group retains its configuration has a barrier of 17.7 kcal/mol and the “forbidden” route where the migrating group inverts its configuration with a barrier of 38.6 kcal/mol (thanks to Alex Genaev; I will strive to make the coordinates available via the repository shortly).
References
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1133933
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1133934
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1133936
- H.S. Rzepa, "Gaussian Job Archive for C5H7B", 2014. https://doi.org/10.6084/m9.figshare.1134015
- M. Menzel, H.J. Winkler, T. Ablelom, D. Steiner, S. Fau, G. Frenking, W. Massa, and A. Berndt, "Diborylcarbenes as Reactive Intermediates in Double 1,2‐Rearrangements with Low Activation Enthalpies", Angewandte Chemie International Edition in English, vol. 34, pp. 1340-1343, 1995. https://doi.org/10.1002/anie.199513401
Tags: Alex Genaev, Hawaii, HB
Posted in pericyclic, reaction mechanism | No Comments »
August 6th, 2014
In my earlier post on the topic, I discussed how inverting the polarity of the C-X bond from X=O to X=Be could flip the stereochemical course of the electrocyclic pericyclic reaction of a divinyl system. An obvious question would be: what happens at the half way stage, ie X=CH2? Well, here is the answer.  The reaction occurs in two stages (ωB97XD/6-311G(d,p)/SCRF=dichloromethane)[1] but overall is a concerted, albeit asynchronous, reaction. The initial stage is a conrotatory ring closure (as observed with X=O but opposite to X=Be), and reaching what we will call a HI (hidden intermediate). This HI clearly has zwitterionic character, and manifests its presence most obviously at IRC = -3.5 below.
The reaction occurs in two stages (ωB97XD/6-311G(d,p)/SCRF=dichloromethane)[1] but overall is a concerted, albeit asynchronous, reaction. The initial stage is a conrotatory ring closure (as observed with X=O but opposite to X=Be), and reaching what we will call a HI (hidden intermediate). This HI clearly has zwitterionic character, and manifests its presence most obviously at IRC = -3.5 below. 
 The polarity of this HI is revealed by the dipole moment (6D) and molecular electrostatic potentials, below. The dipole vector goes from -ve to +ve, and the MEP clearly reveals the polarity below.
The polarity of this HI is revealed by the dipole moment (6D) and molecular electrostatic potentials, below. The dipole vector goes from -ve to +ve, and the MEP clearly reveals the polarity below. 
 This ionic HI however is not stable, and in the second stage of the reaction collapses to the neutral bicyclic hydrocarbon shown below. Overall, it amounts to a 2+2 cycloaddition, but with a very unusual pathway in which one C-C bond is very much formed before the other (which is how the reaction escapes the clutches of the Woodward-Hoffmann forbidden-ness).
This ionic HI however is not stable, and in the second stage of the reaction collapses to the neutral bicyclic hydrocarbon shown below. Overall, it amounts to a 2+2 cycloaddition, but with a very unusual pathway in which one C-C bond is very much formed before the other (which is how the reaction escapes the clutches of the Woodward-Hoffmann forbidden-ness).  Why is all this worth this follow-up? Well, one can now start to “design” the reaction. All three carbon atoms with formal charges can be stabilised or destabilised with appropriate substituents. It should not be too difficult to stabilise out the HI into just an I(intermediate), or indeed to remove it from the profile. Nice perhaps for a group of students, who can partition up the substituents amongst themselves and discover if they have the desired effect. And would any of this tinkering change the stereochemical outcome?
Why is all this worth this follow-up? Well, one can now start to “design” the reaction. All three carbon atoms with formal charges can be stabilised or destabilised with appropriate substituents. It should not be too difficult to stabilise out the HI into just an I(intermediate), or indeed to remove it from the profile. Nice perhaps for a group of students, who can partition up the substituents amongst themselves and discover if they have the desired effect. And would any of this tinkering change the stereochemical outcome?
References
- H.S. Rzepa, "Gaussian Job Archive for C6H8", 2014. https://doi.org/10.6084/m9.figshare.1128205
Tags: Hawaii
Posted in pericyclic | No Comments »
August 6th, 2014
I do go on a lot about the importance of having modern access to data. And so the appearance of this article[1] immediately struck me as important. It is appropriately enough in the new journal Scientific Data. The data contain computed properties at the B3LYP/6-31G(2df,p) level for 133,885 species with up to nine heavy atoms, and the entire data set has its own DOI[2]. The data is generated by subjecting a molecule set to a number of validation protocols, including obtaining relaxed (optimised) geometries at the B3LYP/6-31G(2df,p) level. It would be good to replicate this set with inclusion of a functional that also includes dispersion, and of course making the coordinates all available in this manner greatly facilitates this. The collection also includes data for e.g. 6095 constitutional isomers of C7H10O2, which reminds me of an early, delightfully entitled, article adopting such an approach in quantum chemistry[3]. Such collections are an important part of the process of validating computational methods[4] This way of publishing data does raise some interesting discussion points.
- In this case, we have coordinates for 134 kilo molecules, but the individual molecules in this collection do not have formalised metadata. The InChI key is an example of such metadata, and means that the metadata can be specifically searched. Where you have a monolithic collection of 134k molecules, no such structured exposed metadata exists for individual entries and you will have to generate it yourself in order to search it.
- Each of the molecules in this collection is revealed (once you have downloaded the compressed archive as above and decompressed it into a 548 Mbyte folder) as separate XYZ files.‡ This syntax has the merit of being very simple, and can easily be processed by a human. Computed molecular properties in the form of metadata are missing from this particular (relatively ancient) format. To recover them, you would have to repeat the calculation.
- In fact the XYZ files in this example do seem to have some (unformalised) properties appended to the bottom of the XYZ file (the SMILES and InChI strings are recognizably there, shown as an example below
27
gdb 57483 2.68237 1.10148 0.98017 0.0557 94.95 -0.2958 0.073 ...
C -0.0805964233 1.5844710741 0.1983967506 -0.41097
.........
29.7376 87.1304 196.1576 216.856 ...
CC(C)(C)C1CCCC1 CC(C)(C)C1CCCC1
InChI=1S/C9H18/c1-9(2,3)8-6-4-5-7-8/h8H,4-7H2,1-3H3 InChI=1S/C9H18/c1-9(2,3)8-6-4-5-7-8/h8H,4-7H2,1-3H3
This of itself does raise some issues.
- The title line (starting gdb) has extra numbers, but it is not immediately obvious what these are.
- The XYZ file is no longer standard because extra information is appended, both to each atom line (the charge? shown above as -0.41097) and to the bottom. Much software will not recognise this non-standard XYZ file, and is likely to discard the additional information. Thus I tried wxMacMolPlt (a long time reader of XYZ files) with no success. Human editing of the file was required to remove the additional information before a sensible molecule loaded. Only at this point could one progress to (re)compute the molecular properties.
- The extra information is not formally described. As a human† I can recognise it as an atom coordinate list with appended charges (I think), to which is appended a list of normal coordinate harmonic wavenumbers in units of cm-1, a SMILES and InChI as separate lines. That is really informed guesswork (a human is very good at such pattern recognition) but I cannot be absolutely certain, and a machine seeing this for the first time would certainly struggle.
- The last lines contains repetitions of the SMILES and InChI strings. I am guessing that this is the connectivity determined before and after geometry optimisation (using quantum mechanics, bonds can indeed break or form during such a process) but I may be quite wrong about that. I have not tried to resolve this issue by actually reading the depths of the article, since the file itself really should carry such information.
- The XYZ file itself carries no provenance, such as who created the file, which software and version was used to create it, the date of creation etc.
- An alternative approach is the one adopted here on this blog. Each individual molecule is assigned a DOI and its own metadata and provenance. It is presented to the user in a variety of syntactical forms, each designed for a different purpose, and each adopted for these needs. Thus the syntax and semantics of a CML file are clearly defined by a Schema, and this format can easily absorb additional information without “breaking the standard”. It too can be scaled to 134 kilo molecules[4] although this does require a suitable container (repository) to handle this scale (and I am not entirely sure that DataCite would approve of the generation of 134 kiloDOIs).
Overall, this sort of data publication must be warmly welcomed by the community, and I do hope that more chemistry data is routinely made available in appropriate manner. The presentation in ready-to-reuse form will no doubt improve as the value of such data becomes more fully appreciated. And ultimately, humans need to be excluded from much of this process (editing the 133,885 sets of XYZ coordinates as described above is not for humans to do).
‡Your computer however might balk at opening a folder with 133,885 items in it. Try this only on a very fast machine with lots of memory and ideally an SSD!
†Contrary to some rumors, I do not hail from the planet Zog.
References
- R. Ramakrishnan, P.O. Dral, M. Rupp, and O.A. von Lilienfeld, "Quantum chemistry structures and properties of 134 kilo molecules", Scientific Data, vol. 1, 2014. https://doi.org/10.1038/sdata.2014.22
- Raghunathan Ramakrishnan., P. Dral, P.O. Dral, M. Rupp, and O. Anatole Von Lilienfeld., "Quantum chemistry structures and properties of 134 kilo molecules", 2014. https://doi.org/10.6084/m9.figshare.978904
- P.P. Bera, K.W. Sattelmeyer, M. Saunders, H.F. Schaefer, and P.V.R. Schleyer, "Mindless Chemistry", The Journal of Physical Chemistry A, vol. 110, pp. 4287-4290, 2006. https://doi.org/10.1021/jp057107z
- P. Murray-Rust, H.S. Rzepa, J.J.P. Stewart, and Y. Zhang, "A global resource for computational chemistry", Journal of Molecular Modeling, vol. 11, pp. 532-541, 2005. https://doi.org/10.1007/s00894-005-0278-1
Tags: Much software, validation protocols
Posted in Chemical IT | 5 Comments »